disease. Thus, biopsy of the surrounding flat mucosa should be performed if the patient is symptomatic. However, endoscopists are good at recognizing mucosal pathology grossly in the colon (in contrast to the stomach, in which the endoscopic appearance correlates poorly with the presence of inflammatory processes), and they usually biopsy flat mucosa near a polyp, if the flat mucosa appears abnormal. Before discussing the adenomas and other neoplastic polyps, it is worthwhile to consider some nonneoplastic lesions that produce endoscopic polyps.
TABLE 4.1 Screening Recommendations for Colorectal Cancer and Polyps | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
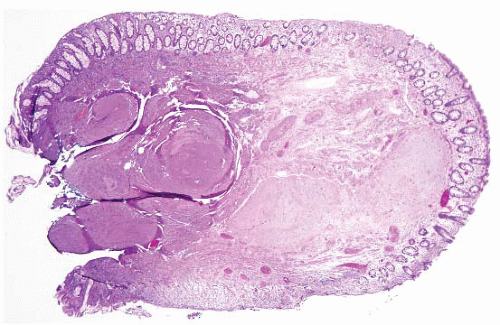 FIGURE 4.1 Colonic elastosis. This example shows a polypoid fragment of tissue with submucosal increase in elastic fibers. |
 FIGURE 4.2 Colonic elastosis. An elastic stain highlights elastic fibers within the submucosa. |
myoglandular polyps are presumably part of the same spectrum (6,7). In some GI sites, prolapse is virtually physiologic. For example, the ileocecal valve is prone to prolapse and, if there is abundant submucosal fat in the prolapsed area, it is termed “lipoma of the ileocecal valve,” but is probably not neoplastic. These conditions are all benign and, occasionally, are mistaken (both clinically and microscopically) for carcinomas.
 FIGURE 4.3 Giant filiform polyposis. Note the fingerlike projections of submucosa covered by mucosa on both sides. This particular case showed impressive active inflammation and presented as a transverse colon mass in a patient without a prior history of IBD. |
peak incidence between 20 and 40 years. The classic history is of a young woman who strains when defecating. There may be hamatochezia, pain, tenesmus, and sometimes lower abdominal pain. Inability to evacuate the rectum, or a “foreign body” sensation, is described. At endoscopy, ulcers are seen in 20% to 70% of patients, usually on the anterior or anterolateral rectal wall, but a mass-like lesion can also be found, which raises the possibility of a neoplasm. Sometimes defecation studies are used to evaluate these patients because they are believed to have difficulty coordinating the smooth muscle during the defecation process, such that the puborectalis sling does not relax at the proper time.
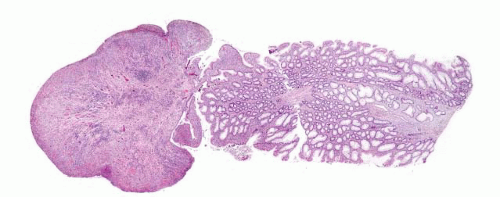 FIGURE 4.4 Filiform polyp. Note the lack of muscularis mucosae in this impressive low power image. |
their results are misleading in the first place). Since prolapse polyps are typically left sided (in contrast to SSA) and feature prominent smooth muscle proliferation, most cases can be assigned to one or the other category. In general, sessile serrated adenomas/polyps lack smooth muscle proliferation in the lamina propria, although some examples have associated perineuriomas/benign fibroblastic polyps (discussed below) in their lamina propria (17-22).
 FIGURE 4.5 Mucosal prolapse. Note fibromuscular stranding into the mucosa with associated surface erosion. |
pattern surface ulceration, displaced clusters of crypts into the submucosa, and abundant fibromuscular stroma that extends into the mucosa.
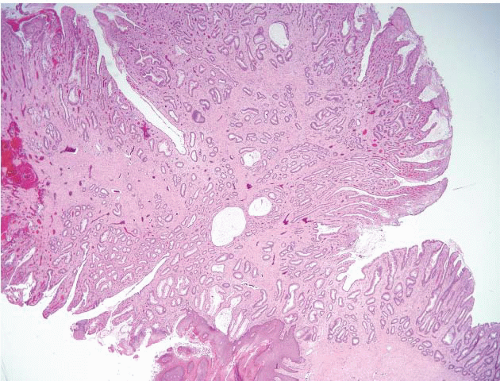 FIGURE 4.6 Inflammatory cloacogenic polyp. This is a mucosal prolapse polyp arising at the anorectal transition. Note both squamous and columnar mucosa, tubulovillous growth pattern, surface ulceration, and abundant fibromuscular stroma that extends into the mucosa. |
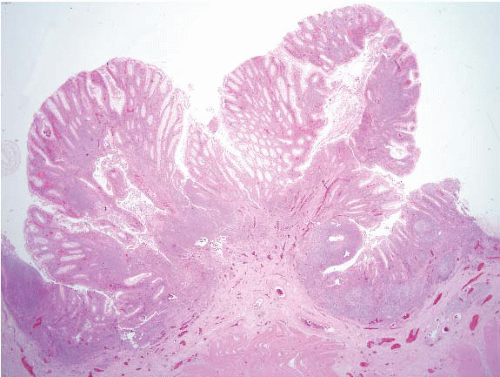 FIGURE 4.7 Diverticular disease associated polyp (polypoid prolapsing mucosal folds). Smooth muscle hypertrophies and contracts around diverticula such that the associated mucosa becomes thrown into redundant folds, prolapsing and forming polyps. The mucosa is commonly inflamed, as seen in this example. |
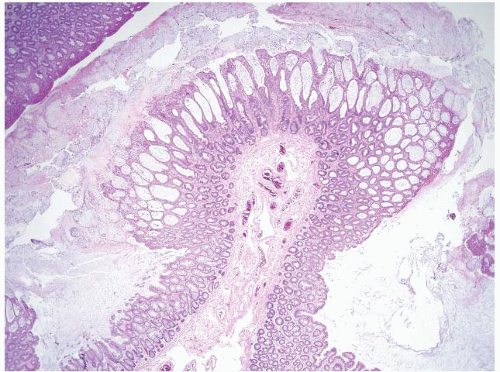 FIGURE 4.8 Cap polyposis. This case shows polypoid hyperplastic epithelium with abundant surface mucus and normal intervening mucosa. |
factor β (TGFβ) superfamily signaling pathways. In Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome, juvenile polyps are a less consistent feature. Cowden syndrome patients are at risk for breast and thyroid cancers. Mutations of the tumor suppressor gene, PTEN, have been found in the germline of both Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome patients. Despite different underlying genetic mechanisms, these and other syndromes share the same phenotypic feature of juvenile polyps. A combined syndrome of juvenile polyposis and hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu disease) has also been described in patients with SMAD4 mutations (54). Hereditary hemorrhagic telangiectasia is an autosomal dominant disorder characterized by vascular malformations of visceral organs usually caused by mutations in ENG (endoglin) or ACVRL1 (ALK1). Though both syndromes have distinct features, a subset of patients with SMAD4 mutations shows a combined phenotype. As a result, patients with juvenile polyposis with SMAD4 mutations should be evaluated for the presence of occult arteriovenous malformations of visceral organs. These syndromes are summarized in Table 4.2.
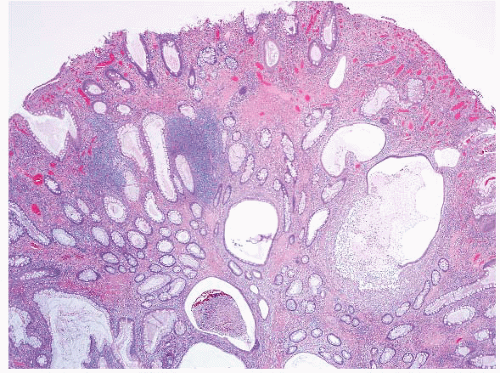 FIGURE 4.9 Juvenile polyp. Note colonic-type mucosa with irregularly shaped and cystically dilated glands with intraluminal acute inflammatory cells, accompanied by an expanded and inflamed lamina propria that contains granulation tissue. |
TABLE 4.2 Summary of Syndromes Associated with Multiple Juvenile Polyps | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
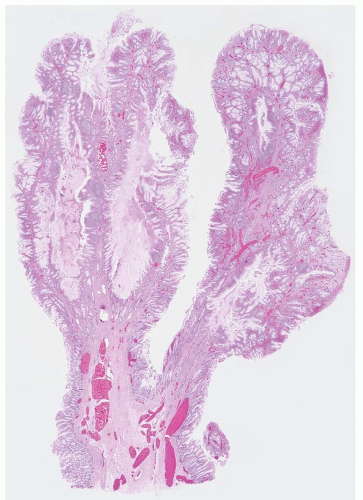 FIGURE 4.10 Syndromic juvenile polyp. Compared to sporadic juvenile polyps, syndromic examples display an increase in the relative amount of epithelium compared to stroma and tend to have fingerlike projections. |
who had undergone colonoscopy, patients had hamartomatous polyps, inflammatory/juvenile-type polyps, ganglioneuromas, adenomas, and two of the 13 had adenocarcinomas (56). Most of the patients had three or more types of polyps in their samples. Examples of polyps encountered in a patient with Cowden syndrome are shown in e-Figures 4.36-4.39.
 FIGURE 4.11 Syndromic juvenile polyp. At high magnification, this juvenile polyp contained extensive low-grade dysplasia (see Figs. 4.31, 4.32 and 4.33) resembling tubular adenoma but the architecture of this polyp is clearly different from that of a sporadic tubular adenoma. Diagnosing such a lesion on a small superficial biopsy would be impossible. |
in the context of clinical history. Overall Peutz-Jeghers polyps differ from prolapse polyps by featuring zones of disorganized mucosa partitioned by cords of smooth muscle, whereas prolapse polyps feature thin strands of smooth muscle replacing lamina propria and investing individual crypts. On small superficial samples, however, it can be impossible to confidently separate the two types of polyps. When we encounter a colon polyp with features of a Peutz-Jeghers-type polyp (Fig. 4.13, e-Figs. 4.44-4.49), we issue a descriptive diagnosis and suggest correlation with other stigmata of Peutz-Jeghers syndrome. Rare examples show dysplasia (e-Fig. 4.50). As discussed in Chapter 3, there are data to suggest that even patients with single “sporadic” Peutz-Jeghers polyps may be at risk for Peutz-Jeghers syndrome-associated malignancies and that isolated examples are the exception (57). Patients with this syndrome are at risk for breast and pancreas cancer, and for female (adenoma malignum of cervix and sex cord tumors with annular tubules of the ovary) and male (sertoli tumors of testis) genital tract tumors. The syndrome associates with mutations/deletions in an involved gene, LKB/STK11, in approximately 80% to 94% of cases.
 FIGURE 4.12 Inflammatory polyp, juvenile type. Some inflammatory polyps in adult patients are presumably a result of prior mucosal injury and can perfectly mimic juvenile polyps. Note the cystically dilated glands and expanded and inflamed lamina propria. |
et al. were able to amass polyps from nine patients from the consultation files of the Armed Forces Institute of Pathology (AFIP) for histologic analysis (59), and Ward has provided excellent reviews of the literature (60-62).
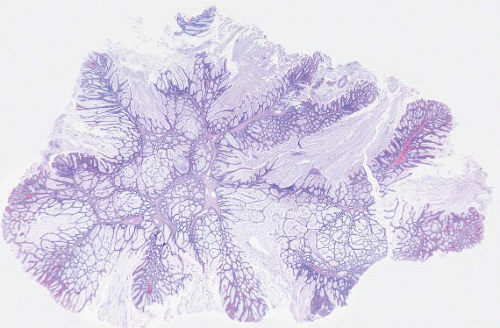 FIGURE 4.13 Peutz-Jeghers polyp. This example shows colonic mucosa partitioned by cords of smooth muscle that invests groups of glands. |
 FIGURE 4.14 Cronkhite-Canada polyp. These are characterized by a broad sessile base, expanded lamina propria, and cystic glands. |
muscularis propria, although deciduosis has rarely been reported in the mucosa of the GI tract. Such cells can sometimes be mistaken for malignant (often epithelial) lesions.
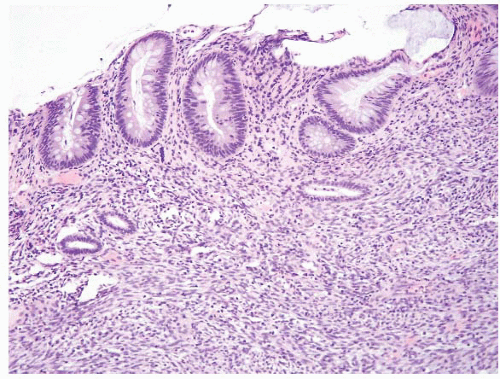 FIGURE 4.15 Colonic endometriosis. This rare example involved the colonic mucosa. Note horizontally oriented endometrial-type glands and associated stroma. |
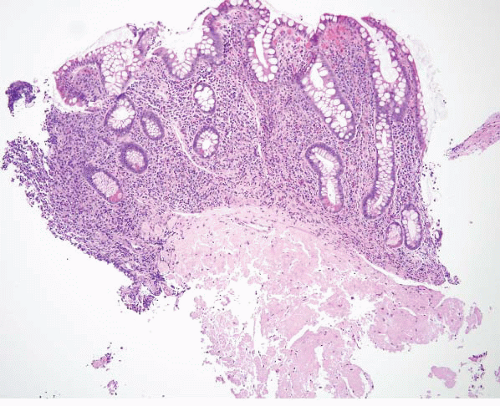 FIGURE 4.16 Amyloidosis. Cases of colonic amyloidosis can give the endoscopic impression of a polyp. Note submucosal amorphous, eosinophilic material. |
the endometrium (67) (Fig. 4.21, e-Figs. 4.84 and 4.85), and Paneth cell differentiation, none of which matter in an adenoma, but which may inform some of the variation in the appearances of invasive carcinomas (66) (e-Figs. 4.86-4.90). Prominent intraepithelial lymphocytes and reduced numbers of
apoptotic bodies are encountered in colorectal adenomas from patients with hereditary nonpolyposis colorectal carcinoma (Lynch syndrome, see below) (e-Fig. 4.91) (68) but it is not known whether finding these features prospectively predicts the diagnosis. Of course, high-grade neuroendocrine (small cell) carcinomas may arise in the background of ordinary-appearing adenomas, but this is rare and discussed below.
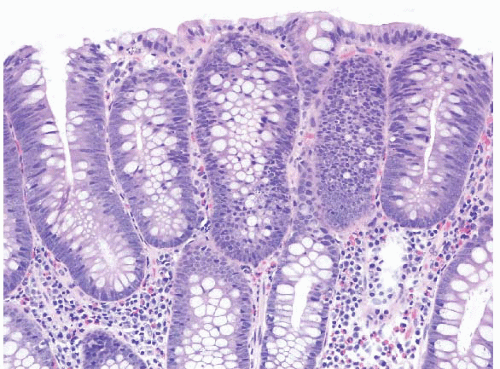 FIGURE 4.17 Tubular adenoma. This example shows low-grade dysplasia with hyperchromatic, pencillate nuclei. Apoptotic bodies are evident in the center of the field. |
 FIGURE 4.18 Tubular adenoma. Note the pencillate, hyperchromatic nuclei that remain perpendicular to the basement membrane. Compare with the normal mucosa on the right, which shows reduced nuclear hyperchromasia and more abundant mucin at the surface. |
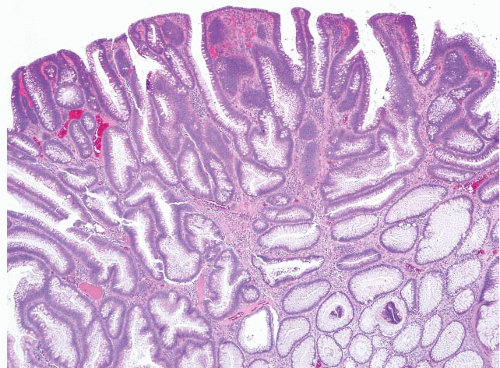 FIGURE 4.19 Tubular adenoma. Some larger examples undergo separation of the crypts at the surface, giving the impression of villi. Tubulovillous adenomas should have long, well-formed fingerlike villi. |
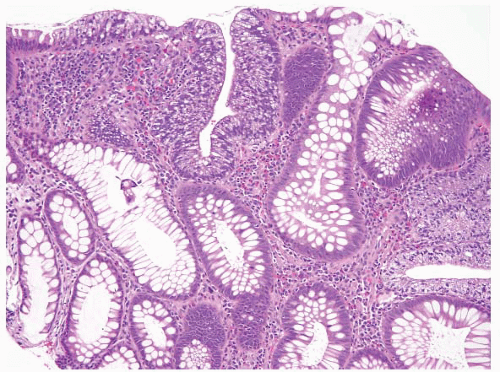 FIGURE 4.20 Tubular adenoma. Some examples contain foci of clear cell change as is seen here in the center of the field. |
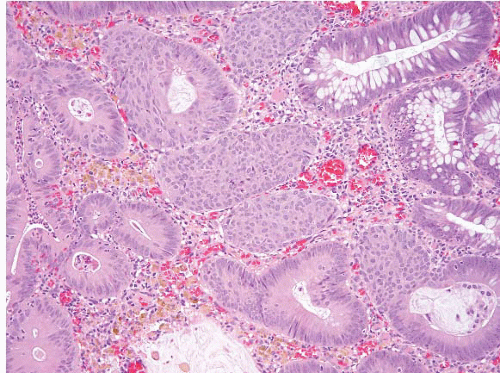 FIGURE 4.21. Tubular adenoma. This example shows squamous-like morules akin to those seen in the endometrium. |
still has no metastatic potential to forestall overly aggressive treatment if the reader of the issued report is not familiar with this basic principle. Criteria to diagnose high-grade dysplasia in colon adenomas are not established, an interesting phenomenon since finding high-grade dysplasia in an adenoma is a reason to intensify post polypectomy follow-up (73).The 2006 guidelines for management after diagnosis of adenomas specifically state “People at increased risk have either three or more adenomas, high-grade dysplasia, villous features, or an adenoma 1 cm or larger in size. It is recommended that they have a 3-year follow-up colonoscopy. People at lower risk who have one or two small (<1 cm) tubular adenomas with no high-grade dysplasia can have a follow-up evaluation in 5 to 10 years, whereas people with hyperplastic polyps (HPs) only should have a 10-year follow-up evaluation, as for average-risk people.” (73). Fortunately high-grade dysplasia and large adenoma size generally go hand in hand, so observer variation in thresholds for high-grade dysplasia are less important than they might seem. Although we have no validation, we generally reserve a diagnosis of high-grade dysplasia in colorectal adenomas for lesions that have cribriform architecture and/or loss of nuclear polarity (Figs. 4.24 and 4.25, e-Figs. 4.103 and 4.104) rather than only cytologic atypia or stratification of nuclei to the surface. The reasoning is that if we miss a bit of intramucosal carcinoma in the colon, it is not of any consequence, whereas we use a lower threshold in the esophagus and stomach. Some colleagues do not even report high-grade dysplasia in colorectal adenomas to forestall overtreatment by surgical colleagues who harbor the erroneous notion that high-grade dysplasia should
prompt a colectomy. When carcinomas arise in adenomas of the colon, invasion of the lamina propria is considered biologically equivalent to highgrade dysplasia (since the lamina propria of the colon is believed to lack lymphatic access, “intramucosal carcinoma” in the colon is thus staged as Tis rather than T1) (74,75), so some observers do not report this invasion either. We report intramucosal carcinoma in adenomas as such and always include a note stating that it is biologically equivalent to high-grade dysplasia (Tis) and that complete polypectomy should be curative. We report our findings this way in case additional sampling discloses deeper invasion.
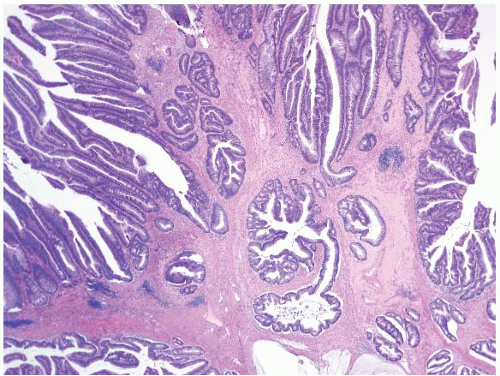 FIGURE 4.22 Pseudoinvasion in a tubular adenoma. Neoplastic glands are seen here hernitated into the submucosa. Note the rounded contours and similar cytoarchitectural features as the adenoma within the mucosa. |
 FIGURE 4.23 Pseudoinvasion in a tubular adenoma. This higher magnification better shows the presence of accompanying lamina propria in the same case as Figure. 4.22. |
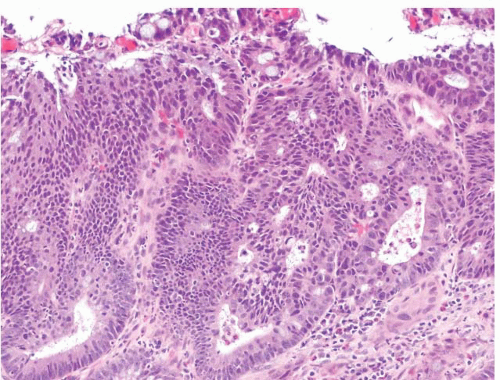 FIGURE 4.24 Adenoma with high-grade dysplasia. Note complex architecture with areas of cribriforming. An area suspicious for lamina propria invasion is seen in the lower right side of the field where cells show more abundant eosinophilic cytoplasm. |
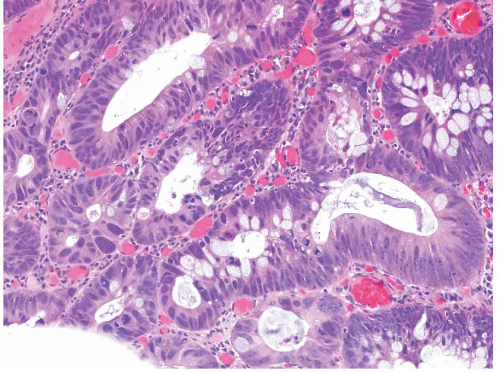 FIGURE 4.25 Adenoma with high-grade dysplasia. This high magnification image shows an area of cribriforming with bizarre cells and loss of nuclear polarity. |
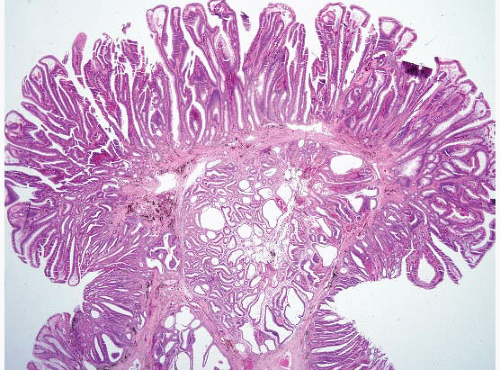 FIGURE 4.26 Tubulovillous adenoma. These polyps show a mixture of both tubular and villous components. Note prominent pseudoinvasion in this example. |
To justify colectomy, the risk of the patient having a metastasis must be higher than the risk of the patient undergoing surgery to remove a segment of the colon. Criteria to make this decision have appeared in the literature since the 1980s and have largely stood the test of time (77,78). Although early studies made a point that there were levels of invasion akin to those in a melanoma (78), other protocols have proved more reliable and more readily assessed in biopsies (79).
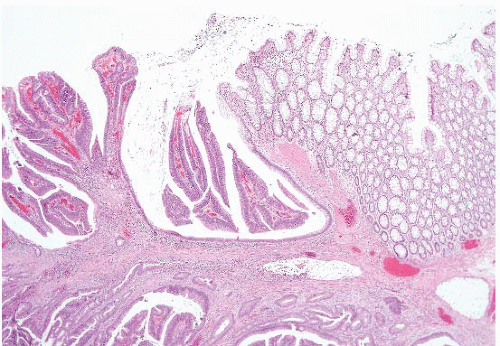 FIGURE 4.27 Villous adenoma. These polyps are composed predominantly of villous architecture characterized by slender, well-formed villi. |
1. A high tumor grade including poorly differentiated adenocarcinoma, signet ring cell carcinoma, small cell carcinoma, or undifferentiated carcinoma (e-Figs. 4.106, 4.107 and 4.114). It remains unclear in the literature whether poorly differentiated carcinomas—that are apparently confined to the lamina propria (e-Fig. 4.115)—have the biologic potential to metastasize. In our study, which included a limited number of patients, polypectomy alone was adequate management for such patients but there are reports of adverse outcomes for patients with intramucosal poorly differentiated colorectal carcinoma (80,81).
2. A tumor ≤1 mm from the resection margin (some authors advise ≤2 mm). We assess this by the not-so-high-tech method of making two small dots (one at the leading edge of the tumor and the other at the nearest cauterized margin) and measuring the distance between them with a ruler. Remember that cauterized tissue contracts, a process that can pull the normal tissue margins together and gives a false impression of positive margins (see e-Fig. 4.112).
3. Involvement of a small (thin-walled) vessel, presumably lymphatic, by the tumor (e-Figs. 4.108-4.110, 4.112, and 4.113).
within an endothelial-lined space. Contraction artifact in the tissue, tumorinduced stromal sclerosis, and extracellular pools of mucin secreted by tumor cells may all complicate the evaluation of vessel invasion. The dilemma may or may not be resolved by the examination of additional tissue levels of the specimen, review by a second observer, and/or immunohistochemical staining for endothelial markers. In published cases in which the malignant polyps have lacked definitive evidence of high-risk features—but the patients have died of their disease—lymphatic invasion had been judged (on blinded review) as indeterminate because of a lack of interobserver agreement (79,82). This suggests that even the suspicion of small vessel invasion on pathologic examination should be considered as potentially important. When there were no adverse features at all, there were no adverse events in this study.
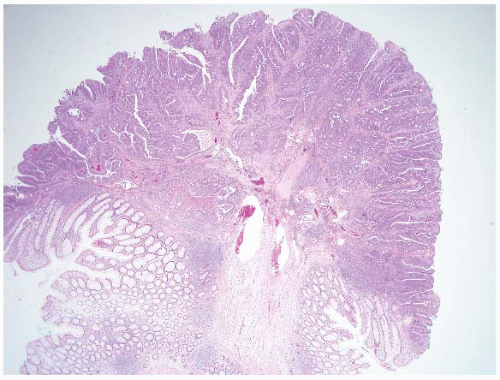 FIGURE 4.28 Invasive adenocarcinoma arising in a tubular adenoma. Malignant polyps are characterized by invasion of malignant glands into the submucosa. Note the adenocarcinoma at the level of large submucosal glands with associated desmoplasia. |
sometimes there can be doubt in samples that do not contain submucosa for evaluation.
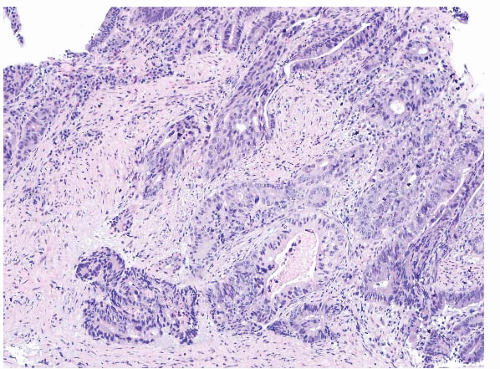 FIGURE 4.29 Colon adenocarcinoma. This biopsy of a colon mass shows angulated glands with central necrosis in a desmoplastic stroma. |
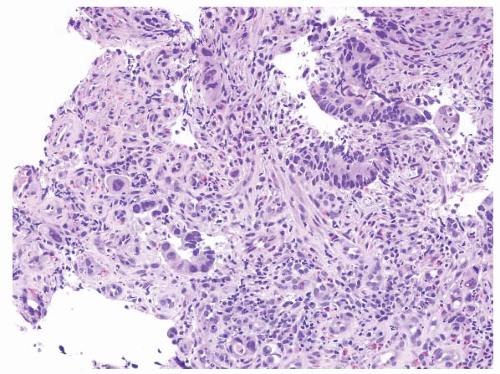 FIGURE 4.30 Rectal adenocarcinoma. This biopsy of a rectal mass shows individual cells and small collections of cells with abundant eosinophilic cytoplasm and prominent nucleoli. |
the lesion is primary (associated with a precursor) and essentially excludes the possibility of a metastasis to the colon.
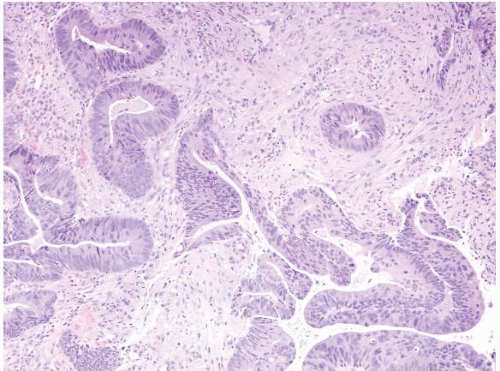 FIGURE 4.31 Rectal adenocarcinoma. This biopsy shows malignant glands associated with well-established desmoplasia. |
TABLE 4.3 Suggested Reflex Testing of Colorectal Cancers | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
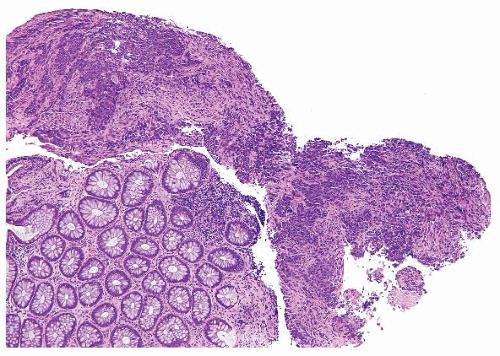 FIGURE 4.32 Colon carcinoma associated with microsatellite instability (MSI). Note the associated lymphocytic infiltrate. |
of MSI. As such, when colorectal cancers are biopsied in a young patient (<50 years), we encourage our clinical colleagues to also sample benign flat mucosa to facilitate this testing, but of course the testing can always be performed later on resected material in which there is abundant normal tissue to use as a control. Electropherograms from both tumor and normal tissues are usually compared when conducting this analysis. The two most commonly used panels are a Promega Microsatellite Analysis System and a reference panel recommended by an NCI-sponsored consensus committee. The Promega system consists of five mononucleotide markers, while the NCI-sponsored reference panel consists of three dinucleotides and two mononucleotides. However, mononucleotide markers show the greatest sensitivity for MSI. MSI is diagnosed when microsatellite lengths are
shifted from the germline pattern. Three possible data interpretations exist: MSI-High (MSI-H), MSI-Low (MSI-L), and Microsatellite Stable (MSS). When five markers are used, a tumor that shows MSI in greater than or equal to two loci is considered MSI-H, one that shows MSI in one locus is termed MSI-L, and one that shows no MSI at any locus is diagnosed MSS.
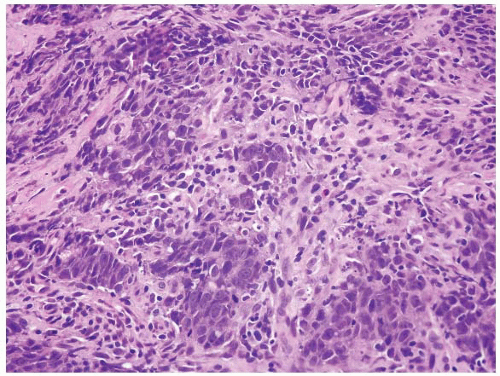 FIGURE 4.33 Colon carcinoma associated with microsatellite instability (MSI). Note the associated lymphocytic infiltrate in this higher magnification of Figure 4.32. |
TABLE 4.4 The Revised Bethesda Guidelines for Testing Colorectal Tumors for MSI | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
for families with a proband that has MSI-H positive tumor, yet who has a negative genetic mutation screen, include counseling the proband and at-risk members as if Lynch Syndrome had been confirmed and beginning a high-risk surveillance program. Patients with MSI-H stage II colorectal cancer have an improved survival relative to other patients. However, it is important to recall that most patients whose tumors have loss of MLH1 protein do not have Lynch syndrome but instead have inactivation of the gene by promoter methylation. However, essentially all patients who show loss of MSH2 by immunostaining (Fig. 4.34) have germline mutations of MSH2. For this reason, we do not perform immunohistochemistry as our first test to evaluate for MMR but instead start with MSI testing. MSI positive tumors can be a result of either germline mutations or sporadic promoter methylation and thus MSI testing does not prove whether a patient has Lynch syndrome. If a patient’s tumor is MSI-H, a discussion can be had after correlation with history as to whether genetic counseling and sequencing of MMR genes is needed. In contrast, if immunolabeling is done reflexively and the patient has loss of MSH2 and the patient has not yet had genetic counseling or given permission for genetic testing, this can be a difficult situation to resolve. Individual institutions need to develop their own protocols for this issue and this testing.
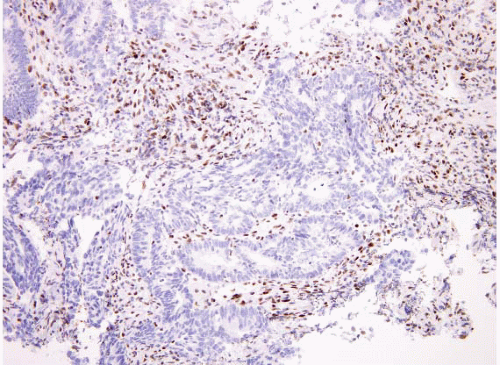 FIGURE 4.34 Colon carcinoma associated with microsatellite instability (MSI). This tumor shows loss of MSH2 by immunohistochemistry. |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


