Chapter 5 Reproductive Genetics
INTRODUCTION
We learn quickly in life that variability exists between individuals. For many of us the differences are as simple as hair or eye color. For others, the differences are profound and can take the form of a severe birth defect or syndrome. As a whole, these differences add up and cause significant morbidity and mortality. A Canadian study has found that approximately 12% of individuals suffer health problems related to or caused by genetic disease from birth to early adulthood.1 Genetics is the study of traits and inherited differences between individuals; as Gregor Mendel demonstrated, we were able to learn the principles of inheritance without knowing about or understanding DNA and its organization in the genome. The traditional study of medicine and medical genetics is, however, about to be revolutionized. In this technological revolution of genomic medicine, we see that a fundamental understanding of genetics and the principles of inheritance are still required.
The practice of clinical medical genetics has been significantly transformed over the past 15 years, from a somewhat arcane specialty dealing with prenatal diagnosis, chromosome abnormalities, and dysmorphology to a challenging and cutting edge specialty taking the lead in introducing genomic medicine to the broader field of medicine. In 2001, the Human Genome Project announced that a rough sequence of the human genome had been completed.2,3 In its original iteration the Human Genome Project was an attempt to sequence and determine the linear sequence of 3 billion base pairs and map the individual genes encoded by this sequence. Genomics is the study of our complete set of genes, their functions and interactions with themselves and their environment, and how genetic variation contributes to disease risk and response to treatment (i.e., pharmacogenomics). This sequencing and mapping effort has now spawned functional genomics, which seeks to identify gene function, regulation, and gene interaction.
The knowledge and technologies learned from this effort have, and will have, as profound an impact on the field of reproductive medicine as any other. The physician in obstetrics and gynecology will not only have to have an understanding of genetics and the principles of inheritance, but he or she will also have to understand genomics and its derivative technologies and how these will affect the risk, diagnosis, and treatment of medical disorders. Physicians will not only be practicing the traditional paradigm of diagnosis and treatment of disease, but will also be recognizing and treating genomically derived disease risk prior to disease manifestation. They must be ready to deal with all the ethical, legal, and social implications of this revolution. Although the era of genomic medicine will bring new tools to individualize disease risk, emphasize prevention, and treat disease, the simple basic recording and understanding of the family history will still be a fundamental component of this new era.4
THE HUMAN GENOME
The Human Genome Project has resulted in some surprising revelations.2,3,5 Less than 2% of the human genome has genes that code for proteins. This was a surprising finding because it was thought that the human genome would reflect the complexity of human development and it was estimated that the human genome would contain up to 120,000 genes. The first report of the Human Genome Project estimated that the human genome contained only 30,000 to 35,000 genes. Drosophila melanogaster had been found to have 14,000 genes and the mustard plant Arabidopsis thaliana 26,000 genes.6,7 It is likely now that the complexity of human growth and development and its relation to disease is not going to be explained by just new gene discovery. The previous dogma that one gene produces one protein has now been replaced by the theory of alternative splicing, in which a gene’s exons or coding regions are shuffled to produce alternative forms of a related protein (Fig. 5-1).
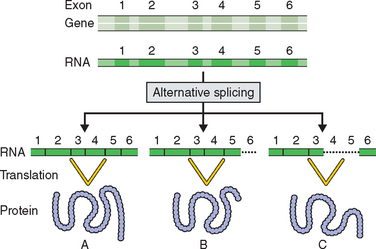
Exons are sequences of a gene that are transcribed into a protein. Introns are regions of a gene that do not code for a protein. When the mRNA of a gene is translated, exons are spliced together. Exons have been found to comprise only 1% of the human genome and introns about 25%. Typically more than 25 times the amount of DNA in a gene is not associated with protein structure and function. This has significant implications for DNA testing and interpretation of a test’s sensitivity and specificity. Regions of genes on chromosomes have been described as existing in gene-rich oases separated by gene-sparse deserts.
Genetic Variation
The human genome contains just over 3 billion base pairs and the sequence is about 99.9% identical in all individuals. It is a surprising finding that human genetic variability may be due to just 0.1% difference in the human genome sequence. A single-nucleotide polymorphism, or SNP, is the most common DNA variant in the genome, with about 10 million occurrences. SNPs are single-base substitutions and occur on average in 1 in every 1250 nucleotides. SNPs are polymorphisms in that a base substitution does not cause a change in the phenotype; they are found in more than 1% of the population. Because of their ubiquity in the human genome SNPs have become invaluable in identifying sequence changes associated with disease risk through traditional linkage studies and population-based association studies. Although not thought to alter protein coding, SNPs have been associated with diseases by their presence in regulatory control regions or introns. SNPs may also determine a disease’s response to treatment, as with the angiotensin-II type 1 receptor polymorphism and its association with congestive heart failure.8
CHROMOSOMAL BASIS OF INHERITANCE
Since Gregor Mendel’s work with the garden pea elucidating the mechanisms of inheritance, genetics has focused on single-gene defects. Modes of inheritance have now been identified for thousands of conditions and catalogued in the online compendium Online Mendelian Inheritance in Man, OMIM at http://www.ncbi.nlm.gov/omim/.
CHROMOSOME ABNORMALITIES
Chromosome Structure
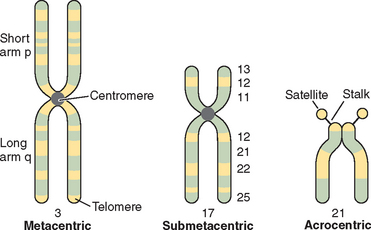
Chromosomes are composed of short arms, p, and long arms, q. The chromosome has a primary constriction, or centromere, where the microtubules attach for cell division. The telomeres, or tips of the chromosomes, are capped with a repeating sequence TTAGGG that is critical for the maintenance of chromosome integrity. The relative position of the centromere further delineates the structure of the chromosome. In humans the acrocentric chromosomes 13, 14, 15, 21, and 22 are characterized by stalks and satellites where genes for ribosomal RNA are located (Fig. 5-2).
Numerical Abnormalities
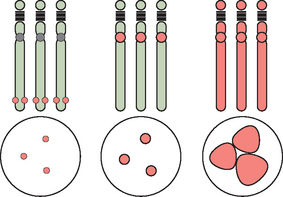
Abnormalities of chromosome number are the most commonly recognized clinical chromosome abnormalities. Structural chromosome anomalies contribute significantly to birth defects, infertility, and recurrent pregnancy loss. Abnormalities in chromosome number generally arise from mistakes at cell division where there is either gain or loss, or both, of chromosomes in daughter cells, or nondisjunction. Nondisjunction can occur either during meiosis or mitosis. Cells resulting from this occurrence are aneuploid because their chromosome number is not a multiple of the haploid number 23. Nondisjunction can occur during either meiosis I or meiosis II. Either can produce an aneuploid conception (Fig. 5-3). Nondisjunction or anaphase lag can occur at mitosis, causing mosaicism, the presence of two or more cell lines in an individual. Mosaicism is often seen with the sex chromosome abnormality Turner’s syndrome, where up to 50% of cases have some form of mosaicism.9 The common numerical chromosome abnormalities are listed in Table 5-1. Numerical chromosome abnormalities can also involve multiples of the haploid number of 23 chromosomes. Triploidy, with 69 chromosomes, usually arises from the fertilization of a single egg by two sperm. Triploid conceptions are seen in about 15% of chromosomally abnormal miscarriages and occasionally survive to term. Tetraploidy has a modal number of 92 chromosomes and occurs in a small percentage of spontaneous losses.
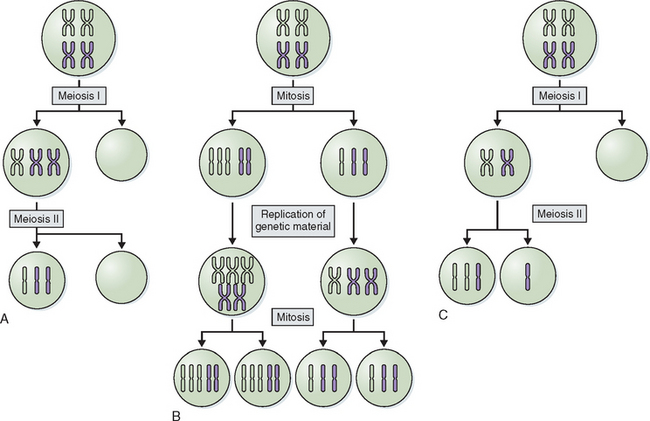
Table 5-1 Chromosome Abnormalities
| Chromosome | Clinical Findings |
|---|---|
| Trisomy 13 | Severe CNS abnormalities, holoprosencephaly, microphthalmia, coloboma, cleft lip and palate, abnormal auricles, polydactyly, cardiac defects |
| Trisomy 18 | CNS malformations, prominent occiput, micrognathia, small mouth, low-set ears, hypoplastic nails, overlapping fingers, cardiac defects |
| Trisomy 21 | Microcephaly, flat occiput, Brushfield spots, epicanthal folds, simian crease, conotruncal cardiac defects, redundant skin on nape of neck, hypotonia |
| XXX | Normal female phenotype, tall stature, may have some learning and developmental disabilities, normal fertility |
| 45,X Turner’s | Short stature, gonadal failure, absent secondary sex characteristics, webbed neck, low-set hairline, coarctation of the aorta, horseshoe kidney, may have some spatial learning disabilities |
| XXY Klinefelter’s | May be tall, infertile, small testis, learning disabilities, gynecomastia |
Structural Chromosome Abnormalities
Structural chromosomal rearrangements occur when chromosomes break and the original architecture is not restored. Chromosome rearrangements are balanced when the diploid genetic state is maintained. When rearrangements are unbalanced, they result in aneuploidy for one or more chromosome segments. These structural rearrangements may segregate and be termed familial, or they may occur as a first or new event, de novo. Balanced familial chromosome rearrangements are in most cases truly balanced and represent little risk of birth defects or mental retardation. De novo chromosome rearrangements that appear balanced carry a small risk of aneuploidy at the molecular level of about 5% for birth defects and developmental delay.
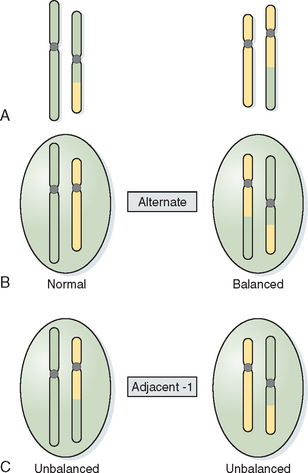
Translocations involve the exchange of chromosome arms between two different chromosomes. Reciprocal translocations occur when there is breakage within two arms and reciprocal exchange of the distal segments, creating a derivative chromosome (Fig. 5-4). In most cases balanced translocation carriers are phenotypically normal but are at risk for producing unbalanced gametes during gametogenesis. In a balanced translocation carrier the types of chromosome segregation can be complex, resulting in a normal segregation pattern, a balanced translocation pattern, and an unbalanced pattern producing a partial trisomy and partial monosomy for the chromosomes involved (see Fig. 5-4).
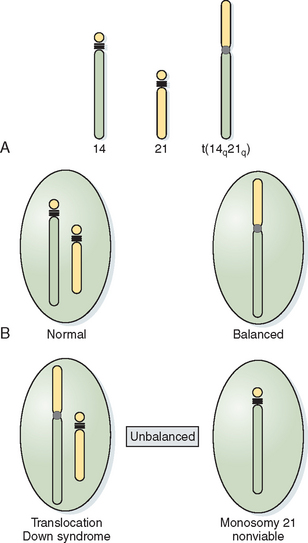
When the short arms of two acrocentric chromosomes are involved in a translocation, the long arms are joined in the centromeric region of one chromosome, with the loss of the short arms of the acrocentric chromosomes producing Robertsonian translocations. Because the short arms of acrocentric chromosomes contain redundant ribosomal genetic material, the loss of this material is of no phenotypic consequence. As with balanced translocations, the products of meiotic segregation can be either balanced or unbalanced (Fig. 5-5).
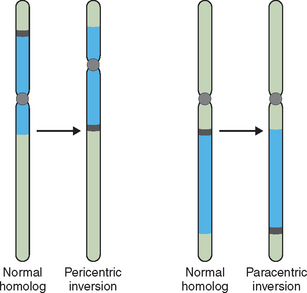
Other structural abnormalities can produce pregnancy loss and birth defects. When two breaks occur in a single chromosome with the interstitial segment flipped 180 degrees at the time of repair, an inversion can occur. If this involves each arm of the chromosome a pericentric inversion is produced. If only a single arm of the chromosome is involved in the inversion, a paracentric inversion is produced (Fig. 5-6). Each has unique and different implications for gamete production and pregnancy loss. With chromosome duplication a chromosome segment of varying size can be duplicated, causing a partial trisomy for this segment. With a deletion, a segment of varying size is missing, causing a genetic imbalance or partial monosomy. This condition, in which a second copy of a gene or segment of chromosome is missing, resulting in an abnormal phenotype or clinical presentation, is known as haploinsufficiency.
Fluorescent In Situ Hybridization
Traditional cytogenetics has always been limited by the band level or resolution of the karyotype. Even with high-resolution banding that could elongate chromosomes and resolve an increasing number of bands per chromosome, one could never be certain that a chromosome was intact at the molecular level. With advances in DNA technology and cytogenetics it is now possible using fluorescent in situ hybridization (FISH) to analyze chromosomes at the molecular level for changes in the DNA. Molecular cytogenetics has now revolutionized cytogenetics by permitting (1) the analysis of DNA structure within a chromosome down to within 10 to 100 kb and (2) the diagnostic analysis of nondividing interphase cells, producing a significant impact on the field of prenatal diagnosis and that of preimplantation genetic diagnosis.10
FISH technology uses DNA probes that can bind or anneal to specific DNA sequences within the chromosome. A denatured probe is incubated with native DNA from a cell that has also been denatured to the single-strand state. The probe substitutes biotin-dUTP or digoxigenin-UTP for thymidine. After the probe has annealed to native DNA, the probe–DNA complex can be detected by adding fluorochrome-tagged avidin that binds to biotin or fluorochrome-labeled antidigoxigenin. This signal can be additionally amplified by adding antiavidin and the complex visualized by fluorescence microscopy. Using several different fluorochromes tagged to different DNA probes, different chromosomes or chromosome segments can be simultaneously visualized within a cell as different colored signals. The ability to detect specific gene segments that are either present or missing has permitted the diagnosis of contiguous gene syndromes at the DNA level as well as translocations in interphase nuclei, often in single cells.
Applications of FISH
FISH technology has been developed in three forms. Centromeric or alpha-satellite probes are relatively chromosome specific and have had probably the broadest application in interphase genetics.11,12 These probes produce somewhat diffuse signals near the centromere with adequate strength, but do not cross-hybridize with chromosomes that have similar centromeric sequences. Single copy probes have now been developed that give discrete signals from a specific band on a chromosome and avoid the issue of cross-hybridization. These can also be used to detect copy number and specific chromosome regions known to be associated with syndromes. Single copy probes and centromeric probes for chromosomes 13, 18, 21, X, and Y have been developed for use in prenatal diagnosis. It is also possible to “paint” whole chromosomes using FISH. Using spectral karyotyping technology that combines mixtures of fluorochromes, it is now possible to produce a unique fluorescent pattern for each individual chromosome in 24 different colors. This technology permits the detection of complex chromosome rearrangements that cannot be seen with traditional cytogenetic techniques (Fig. 5-7).
Prenatal Diagnosis
For an older woman, pregnancy may not be a time of joy but a time of anxiety. Advanced maternal age has a long-known association with an increasing risk of fetal chromosome abnormalities. Amniocentesis done at 16 weeks’ gestation followed by traditional karyotype analysis may take from 10 days to 2 weeks. The use of FISH for preliminary results can expedite a diagnosis and reduce the waiting time for results. Most geneticists and laboratories recommend that FISH not be used alone to make a management decision in pregnancy. FISH studies should always be confirmed with a final karyotype or at minimum correlated with abnormal ultrasound findings or an abnormal maternal serum analyte screen.
Contiguous Gene Syndromes
Contiguous gene syndromes are also known as microdeletion syndromes, or segmental aneusomy.10 These are deletions of a contiguous stretch of chromosome that usually involve multiple genes. The contiguous gene syndromes were first described in 1986 using classical cytogenetic methodologies. Using FISH, submicroscopic deletions can now be identified at the level of DNA; this has permitted the characterization of the smallest deleted region consistently associated with a syndrome, known as the critical region. By identifying the critical region of a syndrome, it is often possible to identify the specific genes that, when missing, are associated with the syndrome (Fig. 5-8). A recent compendium of deletion syndromes has reported 18 deletion and microdeletion syndromes spread over 14 chromosomes.13 Some of the more common deletion and mirodeletion syndromes and their clinical findings are shown in Table 5-2.
Table 5-2 Deletion and Contiguous Gene Syndromes
| Deletion | Syndrome | Clinical Description |
|---|---|---|
| 4p– | Wolf-Hirschhorn | Microcephaly, hypertelorism, frontal bossing, cleft lip and palate, “Greek helmet” face, mental retardation, hypotonia, cardiac anomalies |
| 5p– | Cri du chat | Microcephaly, characteristic “cat cry,” hypotonia, mental retardation |
| 7q11.23 | Williams | Round face, full lips, stellate iris, supravalvular aortic stenosis, mental delay, “cocktail personality” |
| 11p13 | WAGR | Wilm’s tumor, aniridia, genital abnormalities, mental retardation |
| 15q11-13 | Angelman | Blonde hair, prognathism, seizures, ataxia, laughter, hypotonia, mental retardation |
| 15q11-13 | Prader-Willi | Birth hypotonia, hyperphagia, obesity, short stature, hypogonadism, mental retardation |
| 16p13 | Rubinstein-Taybi | Characteristic facies, beaked nose, microcephaly, mental retardation |
| 17p11 | Smith-Magenis | Brachycephaly, prominent chin, short stature, mental retardation, behavioral phenotype |
| 17p13 | Miller-Dieker | Microcephaly, lissencephaly, growth retardation, seizures, mental retardation |
| 20p12 | Alagille | Cholestasis, heart defects, ocular findings, skeletal defects |
| 22q11 | DiGeorge/CATCH 22 | Thymic and parathyroid hypoplasia, calcium abnormalities, conotruncal heart defects, short stature, behavioral and learning problems |
Telomeres
Telomeres are structures that cap the tips of the long and short arms of chromosomes. They are composed of repeating sequences of TTAGGG and effectively prevent the ends of the chromosomes from fusing together. Telomere probes can be important in sorting out complex translocations that cannot be determined by traditional cytogenetic means. In addition, one of the findings of the Human Genome Project was that the chromosome regions next to telomeres are gene-rich. It has now been shown that submicroscopic subtelomeric deletions are responsible for a significant proportion of genetic morbidity.14
GENERAL PRINCIPLES OF MENDELIAN GENETICS
Single-Gene Disorders
In humans as with all diploid organisms, genes come in pairs on related or homologous chromosomes. The exception to this is the sex chromosomes, the X and the Y in the male. Single-gene disorders are described on the basis of the interaction of these pairs or alleles in the genotype and on how the interaction is expressed in the effect or the phenotype. When the alleles are not identical, the genotypes are heterozygous. When the alleles are identical, the genotype is described as homozygous. Gregor Mendel recognized that single-gene disorders are often recognizable and that these single-gene disorders segregate in families with predictable proportions; they have thus been referred to as mendelian traits.15 If a single-gene disorder manifests itself in the heterozygous state, the condition is inherited in a dominant manner. If the single-gene disorder is only inherited when both alleles are affected or altered, the condition is inherited in a recessive fashion. In addition, single-gene disorders are further characterized according to whether they are transmitted on the autosomes or the sex chromosomes.
Autosomal Dominant Inheritance
The following features characterize autosomal dominant inheritance:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


