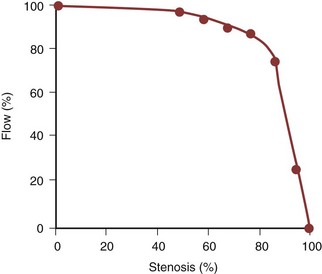
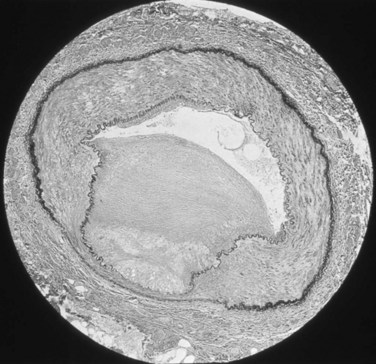
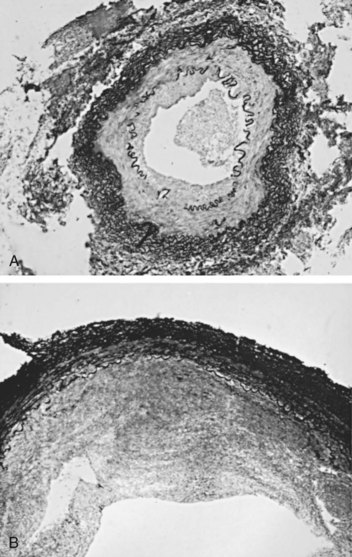
Amr Fergany, MD, PhD, Andrew C. Novick, MD Richard Bright, Physician Extraordinary to the Queen of England, was the first to associate proteinuria, fullness and hardness of the pulse, and dropsy with “hardening of the kidneys” (Bright, 1827). In 1856, Traube, from an analysis of pulse tracings, suggested that the abnormality might be high blood pressure, and Mohomed (1874) demonstrated “high tension in the arterial system” in association with renal disease. The critical experimental work was the discovery of renin by Tigerstedt and Bergemann (1898), who noted an increase in arterial blood pressure in rabbits injected with a saline renal extract. They reasoned that the renal extract contained a pressor substance and coined the term renin. However, the significance of their work was not recognized until the critical experiments by Goldblatt and colleagues (1934), who produced diastolic hypertension in dogs by clamping the main renal arteries and corrected the hypertension by clamp removal. Soon thereafter, Butler (1937) reported the first reversal of hypertension after nephrectomy in a patient with a small “pyelonephritic kidney”; 1 year later, Leadbetter and Burkland (1938) reported another cure of hypertension in a child with pathologic signs of a renal arterial lesion. These clinical observations were paralleled by laboratory investigation, and in 1940, Page and Helmer, and Braun-Menendez and associates, independently reported that renin itself was not a pressor substance but acted as an enzyme to release a pressor peptide, now called angiotensin, from a circulating plasma globulin. Goormaghtigh and Grimson (1939), who had previously described the juxtaglomerular cells, described increased granularity of these cells in both animals and humans with renal hypertension and postulated that these cells were secreting excessive amounts of renin. There followed an aggressive but disappointing clinical experience with nephrectomy for cure of hypertension in patients with unilateral renal disease. This experience led to the search for a way of proving that a renal lesion was actually causing the hypertension. Smith (1948), reviewing the literature, reported relief of hypertension in only 19% of 200 patients whose elevated blood pressure was thought to result from unilateral renal disease. Thus it became apparent that even if pressor mechanisms did underlie some forms of renal hypertension, there were no ways to measure them. This challenge led to studies of the effect of renal artery constriction on renal function. In dogs, renal artery constriction resulted in a marked decrease in sodium and water excretion from the affected kidney (Blake et al, 1950; Pitts and Duggan, 1950). In 1964, Howard and Connor used these observations to develop a differential renal function test based on bilateral ureteral catheterization to identify the “ischemic kidney.” Another major advance was the development of translumbar aortography and the demonstration of its value in visualizing renal arterial lesions (Smith et al, 1952). By 1957, the first large series of studies of patients with renal arterial lesions was reported (Poutasse and Dustan, 1957). In addition, interest in what would become known as the renin-angiotensin-aldosterone system (RAAS) was also emerging as new discoveries were made. Accordingly, it was determined that there were two forms of angiotensin (Skeggs et al, 1954), and angiotensin was sequenced and synthesized (Bumpus et al, 1957). These critical advancements led to an accurate radioimmunoassay for angiotensin, the development of angiotensin analogues, and angiotensin-converting enzyme (ACE) inhibitors, all major tools now used to identify the patient with renovascular hypertension (RVH). More recently, the presence of a family of angiotensin receptors has been clarified (Kang et al, 1994; Goodfriend et al, 1996), and specific blockers of the angiotensin receptor have been used clinically for treatment of hypertension. It is now recognized that the RAAS is a critical integrated system regulating not only blood pressure, sodium balance, and potassium balance but also regional blood flow and, in particular, the glomerular filtration rate (GFR) (Gunning et al, 1996; Laragh and Blumenfeld, 1996). Moreover, there is an expanding body of literature implicating angiotensin II in cell proliferation and interstitial fibrosis (Mai et al, 1993; Eng et al, 1994; Stoll et al, 1995; Egido, 1996; Gunning et al, 1996). As strange as it may seem, it has been difficult to establish a precise definition of hypertension. The problem was best stated by Sir George Pickering, who wrote that “there is no dividing line. The relationship between arterial blood pressure and mortality is quantitative; the higher the pressure the worse the prognosis” (Pickering and Pickering, 1995; Pickering et al, 1996). Indeed, cumulative data obtained from insurance companies have validated this point! Untreated blood pressure in excess of 140/90 mm Hg is associated with excess mortality, and diastolic pressures below 70 mm Hg are optimal (Lew, 1973). For operational purposes, the World Health Organization has defined hypertension in adults as a systolic pressure greater than 160 mm Hg or a diastolic pressure greater than 95 mm Hg or both. In addition, consistent elevation of blood pressure should be established with repeated readings before evaluation is instituted. In children, there is a rise in blood pressure with age; an upper normal limit of 130/80 mm Hg is reached by 12 to 15 years of age. The development of arteriography provided an accurate means of identifying renal arterial disease and heralded the advent of renal arterial vascular repair (Freeman et al, 1954), which renewed enthusiasm for surgical management of the disease. However, it soon became apparent that normotensive patients undergoing arteriography for other reasons often had renal arterial disease (Eyler et al, 1962), especially those with arteriosclerotic disease (Wilms et al, 1990), and autopsy figures supported the radiologic findings (Holley et al, 1964). Accordingly, the finding of renal arterial disease alone is not sufficient justification to warrant correction in a hypertensive patient. The lesion must be functionally significant (i.e., it must reduce blood flow by an amount sufficient to activate renin release, initiating RVH). Hence, a practical definition of RVH is hypertension resulting from a renal arterial lesion that is relieved by correction of the offending lesion or removal of the kidney. The two major pathologic entities that cause renal arterial disease are atherosclerosis obliterans (ASO) and fibrous dysplasia (FD). The Cleveland Clinic group has emphasized the importance of the various distinct histologic patterns, identifiable by angiographic techniques, that have predictable natural histories (Schreiber et al, 1984, 1989; Novick et al, 1994). Their classification is shown in Table 39–1. Table 39–1 Classification and Natural History of Renovascular Disease From Stewart BH, Dustan HP, Kiser WS, et al. Correlation of angiography and natural history in evaluation of patients with renovascular hypertension. J Urol 1970;104:231. Approximately 70% of all renovascular lesions are caused by atherosclerosis (Novick et al, 1996). This disease may be limited to the renal artery but more commonly is a manifestation of generalized atherosclerosis, involving the abdominal aorta and coronary, cerebral, and lower extremity vessels. Atherosclerotic stenosis usually occurs in the proximal 2 cm of the renal artery, and distal arterial or branch involvement is distinctly uncommon. Owing to the proximal location of these lesions, oblique aortic views are often needed to adequately visualize the area of stenosis. The lesion involves the intima of the artery and, in two thirds of the cases, presents as an eccentric plaque (Fig. 39–1); in the remainder, the vessel is circumferentially involved, with narrowing of the lumen and destruction of the intima. Dissecting hematomas frequently complicate this disease, sometimes resulting in thrombosis of the entire vessel. Figure 39–1 Histopathologic appearance of eccentric atherosclerotic plaque causing renal artery stenosis. The natural history of atherosclerotic renal artery disease (RAD) has been studied by obtaining sequential abdominal aortography or duplex ultrasound scanning in patients with documented renal artery lesions who have been treated medically (Table 39–2). The largest of these studies have shown that progressive arterial obstruction occurs in 42% to 53% of patients with atherosclerotic renal artery disease, often within the first 2 years of radiographic follow-up. The incidence of progression to complete renal artery occlusion in these studies has ranged from 9% to 16%, and this has occurred more often in arteries that initially showed high degrees of stenosis. Schreiber and colleagues (1984) reviewed the natural history of atherosclerotic renal artery stenosis (ARAS) in 85 patients who were followed with sequential renal angiograms obtained 3 to 172 months after an initial diagnostic angiogram. Progressive obstruction of the renal artery due to atherosclerosis occurred in 37 patients (44%), including 14 (16%) in whom such progression eventuated in complete occlusion of the involved renal artery. In patients in whom progressive disease developed, it occurred primarily within the first 2 years of angiographic follow-up. The rate of progression of ARAS correlated directly with the degree of stenosis on the initial angiogram. The majority of renal arteries with mild (50%) or moderate (50% to 75%) stenosis on the initial angiogram were unchanged on follow-up angiograms. In contrast, 39% of renal arteries with more than 75% stenosis on the initial angiogram progressed to complete occlusion. Other studies have since validated this observation that progression to 100% occlusion occurs more often and more rapidly in renal arteries that are initially involved with a high degree (>75%) of stenosis (Tollefson and Ernst, 1991; Zierler et al, 1994). Clinical follow-up of patients in the same study (Schreiber et al, 1984) also revealed that significantly more patients with progressive disease developed deterioration of overall renal function compared with patients with stable disease. Interestingly, serial blood pressure control was equivalent in these two groups, indicating that blood pressure is not a useful clinical marker for progressive ARAS. These natural history data clearly show that atherosclerotic renal artery disease progresses in many patients, and that loss of functioning renal parenchyma is a common sequela of such progression. Such loss of renal function due to progressive atherosclerotic renal artery obstruction can result in end-stage renal disease (ESRD). In this setting, ESRD occurs in older patients with generalized atherosclerosis who are not suitable candidates for transplantation and whose prognosis on chronic dialysis is poor in terms of both the quality of life and longevity. An early study identified 25 patients in whom ESRD was clearly a consequence of advanced atherosclerotic renal artery disease (Novick, 1994b). Seventeen of these patients were maintained on chronic dialysis, and, of these, 13 died within 1 year (mean survival 8.7 months). The causes of death on dialysis were myocardial infarction (6), infection (2), gastrointestinal bleeding (1), ruptured aortic aneurysm (1), mesenteric infarct (1), cardiogenic shock (1), and cerebrovascular accident (1). In a subsequent study, Mailloux and colleagues (1988) analyzed the survival of patients started on dialysis from 1970 to 1985 according to the primary renal diagnosis. Patients with renovascular disease as the cause of ESRD had the poorest survival, with a 27-month median survival time and a 12% 5-year survival rate. In another study, in which 51 patients with bilateral ARAS were followed for 52 months, 12% of the patients progressed to ESRD, and an average rate of decline of GFR of 4 mL/min/yr was noted (Baboolal et al, 1998). A crude mortality rate of 45% was reported. These data further highlight that ESRD from atherosclerotic renal artery disease does not respond well to renal replacement therapy. The exact incidence of ESRD caused by atherosclerotic renal artery disease in the United States is not known. Fatica and colleagues (2001) reported an increase in incidence of renal vascular disease (RVD) as a cause for ESRD in patients starting dialysis treatment. This increase was from 1.4% to 2.1%, with an annual increase of 12%. This information was derived from the recorded diagnosis of these patients in the U.S. Renal Data System database, and the disease was not specifically searched for. No increase in mortality on dialysis was found in these patients when compared with other etiologies of ESRD. When investigating the use of CT angiography, van Ampting and coworkers (2003) reported a 27% incidence of significant renal artery stenosis (RAS) in 49 patients over 45 years of age starting dialysis. Uzu and coworkers (2002) reported a higher (50%) incidence in 44 patients with ESRD who were studied by magnetic resonance angiography when additional vascular disease (cerebral, coronary, or peripheral) was also diagnosed. In a report from England, Scoble and colleagues (1989) prospectively performed renal arteriography in all new patients with ESRD during an 18-month period. Atherosclerotic renal artery disease was the cause of ESRD in 6% of all patients, and in 14% of patients older than 50 years. Approximately 300,000 patients in the United States are currently being maintained on chronic dialysis. Their median age is older than 60 years, and a majority show evidence of generalized atherosclerosis obliterans. Although the exact number of patients with ESRD caused by atherosclerotic renal artery disease is not known, based on the data previously described, there appear to be several thousand patients in this category. Primary intimal fibroplasia occurs in children and in young adults and constitutes approximately 10% of the total number of fibrous lesions. This lesion is characterized by a circumferential accumulation of collagen inside the internal elastic lamina (Fig. 39–2). Disruption and duplication of the elastica interna occur more often in younger patients, with dissecting hematomas as a complication in many patients. The possibility of atherosclerosis as a cause of renal artery disease in this group can be excluded histologically by the absence of lipid demonstrable with special staining techniques. Intimal fibroplasia with complicating medial dissection is characterized pathologically by large dissecting channels in the outer half of the media. These lesions are thought to develop because of defects in the internal elastica with resultant medial dissection and aneurysmal dilatation. (From Novick AC. Renal vascular hypertension in children. In: Kelalis PP, King LR, Belman AB, editors. Clinical pediatric urology. Philadelphia: WB Saunders; 1984.) Angiography in primary intimal fibroplasia reveals a smooth, fairly focal stenosis, usually involving the proximal or midportion of the vessel or its branches (Fig. 39–3). Dissecting hematomas may distort the area of the stenosis. With nonoperative management, progressive renal artery obstruction and ischemic atrophy of the involved kidney invariably occur. Severe intimal fibroplasia may subsequently develop de novo in the contralateral renal artery. Although primary intimal fibroplasia most commonly affects the renal arteries, it may also occur as a generalized disorder with concomitant involvement of carotid, upper and lower extremity, and mesenteric vessels. (From Novick AC. Renal vascular hypertension in children. In: Kelalis PP, King LR, Belman AB, editors. Clinical pediatric urology. Philadelphia: WB Saunders; 1984.) Angiographically, medial fibroplasia demonstrates a typical “string of beads” appearance involving the distal two thirds of the main renal artery and branches (Fig. 39–4). The areas of stenosis are often overshadowed by contrast medium in the microaneurysms, making the degree of actual stenosis difficult to assess. The aneurysms themselves are greater in diameter than the normal renal artery proximal to the disease, and extreme collateral circulation is absent. These are important features in differentiating the lesion from perimedial fibroplasia. Schreiber and colleagues (1984) studied the natural history of renal artery disease due to medial fibroplasia in 66 patients who were followed with serial angiography. Progressive renal artery stenosis (RAS) occurred in 22 patients (33%), and, contrary to an earlier report, this occurrence was no different whether patients were older or younger than 40 years. Significantly, there were no cases of progression to total arterial occlusion in this group. Also, clinical follow-up revealed that serial decreases in either overall renal function or the size of the involved kidney seldom occurred in patients with progressive medial fibroplasia, suggesting that the risk of losing renal function is relatively small in patients with this disease who are managed medically. (From Novick AC. Renal vascular hypertension in children. In: Kelalis PP, King LR, Belman AB, editors. Clinical pediatric urology. Philadelphia: WB Saunders; 1984.) Perimedial fibroplasia occurs predominantly in young women between the ages of 15 and 30 years and has therefore been referred to, rather crudely, as girlie disease. It constitutes about 10% to 15% of the total number of fibrous lesions and occurs only in the renal artery. This is a tightly stenotic lesion that, pathologically, consists of a collar of dense collagen enveloping the renal artery for variable lengths and thicknesses. The collagen is deposited in the outer border of the media, usually replaces a considerable portion of the media, and may replace it completely in some areas (Fig. 39–5). Islands of smooth muscle are occasionally seen trapped within the collagenous ring. Special stains show that the lesion is confined within the external elastic lamina and contained in all cases by intact adventitial connective tissue. The arterial lumen may be further compromised by a process of secondary intimal fibroplasia. It has been suggested that this secondary thickening of the intima is related to slowing of blood flow through a narrowed arterial segment, with resultant platelet and fibrin deposition and subsequent fibrous organization. (From Novick AC. Renal vascular hypertension in children. In: Kelalis PP, King LR, Belman AB, editors. Clinical pediatric urology. Philadelphia: WB Saunders; 1984.) The arteriogram in perimedial fibroplasia may give the appearance of arterial beading, but careful observation shows that the caliber of the normal segment of the vessel is not exceeded by the “bead” (Fig. 39–6). This fact, along with the frequent occurrence of extensive collateral circulation, differentiates this lesion angiographically from that of medial fibroplasia. Perimedial fibroplasia produces severe stenosis, and, although complicating thrombosis or dissection is relatively uncommon, progressive obstruction with ischemic renal atrophy occurs in almost all patients managed nonoperatively. (From Novick AC. Renal vascular hypertension in children. In: Kelalis PP, King LR, Belman AB, editors. Clinical pediatric urology. Philadelphia: WB Saunders; 1984.) Key Points: Pathology of Renal Artery Stenosis Angiotensinogen is a 452–amino acid protein and is the source of all angiotensins (Kageyama et al, 1984). It is formed as preangiotensinogen and loses the signal peptide as it becomes secreted from the cell as angiotensinogen. It functions as a serine protease inhibitor (serpin) similar to α1-antitrypsin and antithrombin III, with which it shares some structural homology (Carrell et al, 1987). It is present in plasma in two forms, a smaller (52- to 60-kD) predominant molecule and a larger (450- to 500-kD) molecule that increases in pregnancy and after estrogen treatment (Tewksbury and Dart, 1982). The larger form is probably composed of the smaller molecule bound to other plasma proteins. Renin acts on the smaller form of angiotensinogen preferentially, cleaving AI off the larger molecule. Renin reacts with much less affinity with the larger form, also forming AI. The liver is the primary site of synthesis of angiotensinogen, which is not stored but secreted directly after production. Angiotensinogen mRNA is widely present in several tissues that are regulated by local renin-angiotensin systems, including the central nervous system (CNS), kidney, adrenal, heart, and leukocytes (Dzau et al, 1987). Several hormones stimulate angiotensinogen synthesis by the liver, including estrogens and glucocorticoids. Stressful stimuli, such as infection or tissue injury, also increase plasma angiotensinogen levels (Hoj Nielsen and Knudsen, 1987). Feedback control through the RAAS is also present, with AII increasing and renin decreasing plasma levels of angiotensinogen. Renin is a single–polypeptide chain aspartyl protease that is secreted from the juxtaglomerular cells of the afferent arteriole. The kidney is the major site of renin production, although renin mRNA is found in several other tissues where a local renin-angiotensin system functions. It is produced as pre-prorenin, and both active renin and prorenin are secreted (Atlas et al, 1980). The function of circulating prorenin is not clear, and it does not appear that prorenin is transformed to active renin in the circulation (Sealey et al, 1977). The action of renin is very specific, restricted to cleavage of a single bond, separating AI from angiotensinogen. Because renin controls the rate-limiting step of the RAAS, control of renin secretion regulates the activity of the RAAS. Several mechanisms affect the secretion of renin, as described in the following sections. As the first and rate-limiting step for production of angiotensin II, targeting renin is an attractive option for inhibiting the RAAS. Recently a new class of orally effective medications targeting renin has been developed and approved for treatment of hypertension, direct renin inhibitors (DRI). The first of these medications is aliskiren, is a competitive analog and specific inhibitor of human renin, with therapeutic potential similar to other available antagonists of the RAAS (ACE inhibitors, angiotensin receptor blockers) (Nussberger et al, 2002). The macula densa region of the thick ascending loop of Henle comes in close proximity to the juxtaglomerular cells and influences renin release. Reduction of distal tubule salt delivery stimulates renin secretion, and vice versa. Although sodium was initially thought to be responsible for this action, it now appears that the signal for macula densa–controlled renin release is the alteration of tubular chloride concentration (Lorenz et al, 1990). The juxtaglomerular cells of the afferent arteriole act as their own baroreceptors by responding directly to stretch of the afferent arteriole (Tobian et al, 1959). Diminished cell stretch, as a result of renal hypoperfusion, hyperpolarizes the juxtaglomerular cells, resulting in decreased intracellular calcium and increased renin release. The juxtaglomerular cells are richly innervated by β-adrenergic sympathetic nerve fibers. Stimulation of these β-adrenergic nerves leads to increased renin secretion (Keeton and Campbell, 1980). Dopamine is also stimulatory to renin release, although the limited number of dopaminergic nerve endings results in a much smaller role (Mizoguchi et al, 1983). Renal nerve stimulation is the mechanism through which renin release is increased as a result of exercise and tilting. Several local and systemic hormones affect the rate of renin secretion. Foremost among these are prostaglandins. Prostaglandin E2 and I2 (prostacyclin), as well as exogenously administrated arachidonic acid, stimulate renin secretion (Franco-Saenz et al, 1980; Whorton et al, 1980). This prostaglandin effect is independent of the other mechanisms controlling renin release. AII inhibits renin release as a feedback mechanism. Other inhibitors of renin release include endothelin, vasopressin, and atrial natriuretic peptide. ACE is a zinc-containing single-chain glycoprotein enzyme. It is also known as kininase II and is a dipeptidyl carboxypeptidase (Ehlers and Riordan, 1989). It splits two amino acids off the carboxy terminus of AI to form AII and, at the same time, functions in the kallikrein-kinin system by inactivating bradykinin. ACE is found in a wide variety of organs, where it is primarily expressed on endothelial, epithelial, and neuroepithelial cells. A high concentration of ACE is found in the kidney, ileum, duodenum, and uterus (Lieberman and Sastre, 1983). Although pulmonary endothelial ACE was presumed to be the major site of ACE activity for the systemic RAAS, it is now believed that peripheral sites might play an equal role. The majority of circulating ACE originates from endothelial cells and macrophages. ACE is expressed in several tissues where local renin-angiotensin systems function. Renal ACE is localized to the glomerular endothelial cells and the proximal tubule brush border, where it might play a role in cleaving filtered protein for reabsorption (Danilov et al, 1987). Within the CNS, ACE is found in several locations, where it functions in the local renin-angiotensin system. This local CNS renin-angiotensin system is thought to have dipsogenic and hypertensive effects as well as to stimulate vasopressin secretion (Strittmatter and Snyder, 1987). Adrenal ACE is found predominantly in the medulla, where it is thought to stimulate catecholamine secretion (Peach et al, 1971). ACE is found abundantly in the testes and prostate, in the Leydig cells, and also in cytoplasmic droplets in sperm (Pandey et al, 1984; Yotsumoto et al, 1984). In the female reproductive tract, ACE is found in follicular and fallopian tube oocytes (Brentjens et al, 1986). The precise role of ACE in the reproductive system has not been elucidated. Several hormones and disease states affect the level and activity of ACE. Corticosteroids, as well as thyroid hormones, stimulate ACE activity (Friedland et al, 1978; Smallridge et al, 1983). The serum ACE level is increased in silicosis, primary biliary cirrhosis, and sarcoidosis (Studdy et al, 1983). As mentioned previously, ACE is not the rate-limiting step in the RAAS cascade; so, changes in serum ACE levels do not directly affect the activity of the systemic RAAS (circulating AII levels). One of the most important actions of AII is the autoregulation of the GFR in response to changes in renal perfusion. These are affected through changes in vascular resistance as well as mesangial cell tone. AII causes a marked increase in efferent arteriolar resistance in cases of renal hypoperfusion but does not affect afferent arteriolar resistance unless there is an increase in renal perfusion pressure. The result of this disproportionate increase in efferent over afferent resistance is an increase in capillary hydraulic pressure, and subsequently in filtration pressure, maintaining the GFR in the face of decreased renal perfusion (Hall et al, 1977). It is through inhibition of this action that ACE inhibitors result in a decrease in GFR in cases of renal artery stenosis. This effect of AII on the glomerular circulation is thought to be mediated through differential induction of vasodilatory prostaglandins from the afferent and efferent vessels (Hura and Kunau, 1988). In addition to its effects on the glomerular vessels, AII directly results in mesangial cell contraction, leading to a decrease in the filtration coefficient of the glomerulus (Blantz et al, 1976). AII-induced increases in the filtration fraction lead to an increase in the oncotic pressure in the postglomerular vessels. This leads to an increase in fluid reabsorption in the proximal tubules. AII receptors are also present on the proximal tubule brush border and basolateral sides, and AII is produced in large amounts locally within the proximal tubules. AII is present within the proximal renal tubule in much higher concentration than in the plasma (Seikaly et al, 1990). The effect of AII on sodium reabsorption is bimodal; physiologic concentrations of AII stimulate sodium reabsorption in the proximal tubule, whereas higher concentrations inhibit sodium transport (Harris and Young, 1977). AII decreases medullary blood flow, leading to increased medullary hypertonicity and concentration of urine (Arendshorst and Finn, 1977). AII raises blood pressure by increasing peripheral vascular resistance through a direct effect on vascular smooth muscle cells, causing them to contract. Medium-sized and small arteries are more responsive to AII than large vessels. Contraction occurs mainly in the vessels of the kidney, skin, mesentery, coronary arteries, and brain. Vessels of the lung and skeletal muscle are less responsive to AII. In addition to vasoconstriction, AII stimulates vascular smooth muscle cell growth, leading to a hypertrophic response (Geisterfer et al, 1988). Such smooth muscle proliferation results in left ventricular hypertrophy in cases of chronic stimulation of the RAS. AII is also involved in inflammatory processes including atherosclerosis. This cascade of cardiovascular events including hypertension, ventricular hypertophy, and atherosclerosis is felt to be central to the development of heart failure. With this mechanism in mind, interruption of the RAAS through multiple pharmacologic channels has become a major objective in lowering cardiac mortality associated with hypertension and heart failure. AII acts directly on the adrenal glomerulosa cells to stimulate aldosterone secretion. This is accomplished through increased desmolase activity and increased conversion of corticosterone to aldosterone (Aguilera, 1993). This augments the salt reabsorptive actions of AII to conserve sodium. The CNS is affected mainly by the local renin-angiotensin system, but high circulating levels of AII may also affect CNS function. Central AII results in an increase in blood pressure as well as increased drinking and salt appetite (Sweet et al, 1971; Fitzsimons, 1980). Central AII also leads to increased secretion of corticotropin, prolactin, luteinizing hormone, oxytocin, and vasopressin (Unger et al, 1988). Nonpeptide receptor antagonists have provided definite proof of at least two major angiotensin receptor subtypes, named AT1 and AT2. Both receptors are polypeptides containing 360 amino acids spanning the cell membrane several times. They are functionally distinct with a sequence homology of 30%. The gene for the AT1 receptor is located on chromosome 3, and the gene for the AT2 receptor is located on the X chromosome (Goodfriend et al, 1996). AT1 receptors are blocked by DuP 753 (losartan), and AT2 receptors are blocked by tetrahydroimidazopyridines such as PD 123177. AT1 receptors have a higher affinity for AII than AIII, but AT2 receptors bind both AII and AIII equally. AT1 receptors have been further subtyped into two isoforms, AT1A and AT1B, although the function of the subtypes is not clear. In the kidneys, AT1 receptors are located predominantly in the glomeruli and tubulointerstitium, whereas AT2 receptors are located in the large cortical blood vessels (Goldfarb et al, 1994). Almost all the vascular effects of AII, including vasoconstriction, aldosterone release, and β-adrenergic stimulation, are mediated by the AT1 receptor (Timmermans et al, 1992). The development of AT1 receptor antagonists (e.g., losartan) has produced a new class of drugs, as well as an effective tool for blocking the RAAS in a variety of disease states, including hypertension. Further, these new drugs modulate cardiac and renal injury responses to disease. The function of the AT2 receptor has not been fully defined; however, it may act in a manner antagonistic to the AT1 receptor, especially in the cardiovascular system, where it exerts antiproliferative, antihypertrophic, and proapoptotic functions (Horiuchi et al, 1999). AT2 receptors are thus thought to mediate protective actions that counterbalance the potentially harmful actions mediated through the AT1 receptors. AT2 receptors are also believed to play a crucial role during gestational growth and development, mainly due to the widespread distribution of these receptors in most body tissues during fetal life. Reexpression of these receptors in adult life occurs as a response to vascular injury or inflammation (Horiuchi et al, 1999). The signal transduction mechanism initiated by binding of AII to the AT1 receptor has been well described; however, the signal transduction mechanism for the AT2 receptor is not yet known. Binding of AII to the AT1 receptor leads to the dissociation of subunits of a guanine nucleotide–binding protein, which activates phospholipase C to generate diacylglycerol and inositol triphosphate. Inositol triphosphate releases calcium from the endoplasmic reticulum, and AII also increases calcium entry through the cell membrane. The intracellular calcium, as well as diacylglycerol, activates protein kinase C and other enzymes that phosphorylate protein and ultimately regulates the specific cellular function induced by AII (Goodfriend et al, 1996). The parent peptide of the angiotensin family is the decapeptide AI. Several other peptides are formed within the RAAS, some of which have weak activity compared with AII, and some of which have undetermined activity. As previously mentioned, AII (also called angiotensin 1-8) is the major active peptide in the system; it is an octapeptide formed by the removal of terminal histidine and leucine from the carboxy terminus of AI. AIII (or angiotensin 2-8) is similar to AII but lacks the aspartyl amino acid at the amino terminus of the polypeptide chain. It can be formed from AII or directly from AI. Angiotensin 1-7 lacks the three amino acids at the carboxy terminus of AI and has undetermined receptor activity. AIV is a hexapeptide lacking the two terminal amino acids at both ends of the AI polypeptide chain (Goodfriend et al, 1996). The actions of angiotensin 1-7 have been defined. It appears to be formed from AI directly by a different enzyme than ACE, called neprilysin. ACE inhibitors thus increase the levels of circulating angiotensin 1-7. It acts in an opposing fashion to AII, producing vasodilatation and natriuresis, and also has antiproliferative effects on vascular smooth muscle (Chappell and Ferrario, 1999). The classic experiments on RVH were performed by Goldblatt and colleagues (1934), who demonstrated that hypertension could be produced by constricting the renal artery in the dog. Two models of experimental Goldblatt hypertension are described: the two-kidney, one-clip (2K,1C) model, in which one renal artery is clipped and the contralateral kidney is in place and normal; and the one-kidney, one-clip (1K,1C) model, in which one renal artery is clipped and the contralateral kidney is removed. RVH results in both models, but the evolution and the pathophysiologic mechanisms are different. These models provide the basis for understanding the mechanism and evolution of RVH in humans. In addition, both models do not remain static but, rather, pass through an acute phase, a transitional phase, and then a final chronic phase (Table 39–3). In cases of two-kidney, one-clip hypertension, after several days or weeks a chronic phase is eventually reached in which unclipping of the stenotic kidney fails to normalize blood pressure. In this chronic phase, the elevated perfusion pressure, as well as high levels of AII, results in widespread arteriolar damage to the contralateral kidney. The excretory function (natriuresis) of the contralateral kidney declines, resulting in extracellular volume expansion, a decrease in circulating AII levels, and the gradual development of a “volume-dependent” type of hypertension. ACE inhibition or removal of the stenotic kidney fails to cure the hypertension in this phase of the disease unless sodium depletion is instituted. Systemic vasoconstriction continues to play a role in maintaining hypertension in the chronic phase, with increased sensitivity to AII, increased vasopressin secretion, and increased sympathetic nervous system activity. Table 39–3 Phases of Experimental Renovascular Hypertension IN is the result of chronic hypoperfusion of the total functioning renal mass. This occurs in the setting of bilateral severe stenosis or stenosis to a functionally or anatomically solitary kidney. The pathophysiology of renal injury as a result of chronic ischemia is poorly understood. This injury is not simply cell death due to a lack of oxygen and nutrients, because the oxygen demand of the kidney never exceeds the supply. Experiments studying the effects of acute renal ischemia do not lend themselves to the explanation of chronic ischemic injury. For ischemic injury to occur, the reduction in renal blood flow needs to exceed the compensatory ability of the kidneys. Renal autoregulation fails to maintain the GFR when renal perfusion decreases below 70 to 80 mm Hg. This occurs when the luminal diameter of the renal artery is stenosed by more than 70% of the original size. At this point, the stenosis becomes hemodynamically significant, resulting in a gradual deterioration of the GFR with an accompanying rise in the serum creatinine level (Fig. 39–7).
Historical Background
Definitions
Hypertension
Renal Arterial Disease versus Renovascular Hypertension
Pathology and Natural History
Atherosclerosis: Proximal intimal plaques. Seen predominantly in males and usually in older age groups. Progressive in about 40% of patients; may dissect or thrombose. May involve renal arteries only or may involve carotid and coronary arteries, aorta, and other vessels.
Intimal fibroplasia: Collagenous disease involving intima; seen in children and young adults. Progressive; may dissect. May involve other vessels.
True fibromuscular hyperplasia: Diffusely involves media. Seen in children and young adults. Progressive. Radiographically indistinguishable from intimal fibroplasia. Very rare.
Medial fibroplasia: Series of collagenous rings involving media of main renal artery, often extending into branches. Usually seen in women in their 30s and 40s. Produces typical “string of beads” pattern in angiography. Does not dissect, thrombose, or rupture, and seldom progresses after 40 years of age. May involve other vessels.
Perimedial (subadventitial) fibroplasia: Dense collagenous collar involving media, just beneath adventitia of vessel. Tightly stenotic, with extensive collateral circulation on angiography. Seen mostly in women (“girlie disease”). Progressive. Involves renal arteries only.
Miscellaneous: Renal artery aneurysms, middle aortic syndrome, periarterial fibrosis, and post-traumatic intimal or medial disease. Variable in location and obstruction; occurs in diverse clinical settings.
Atherosclerosis
Fibrous (Fibromuscular) Dysplasia
Intimal Fibroplasia
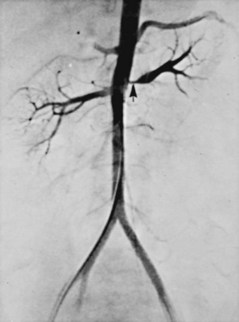
Medial Fibroplasia
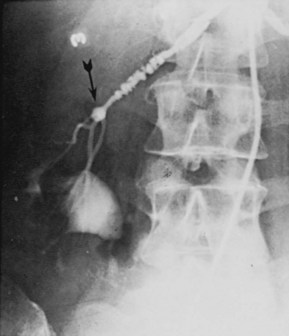
Perimedial Fibroplasia
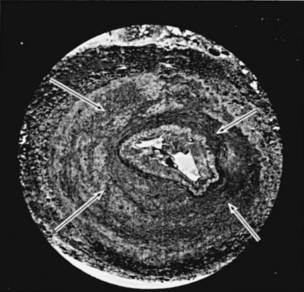
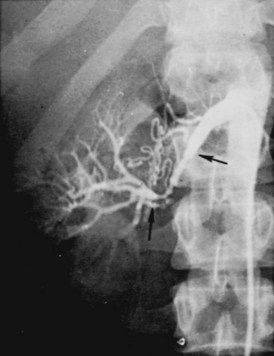
Fibromuscular Hyperplasia
Physiology of the Renin-Angiotensin-Aldosterone System
Angiotensinogen
Renin
Macula Densa Mechanism
Baroreceptor Mechanism
Neural Mechanism
Endocrine and Paracrine Mechanisms
Angiotensin-Converting Enzyme
Effect of Angiotensin II on Glomerular Circulation
Tubular Effects of Angiotensin II
Medullary Effects
Vascular Effects
Adrenal Effects
CNS Renin-Angiotensin-Aldosterone System
Angiotensin II Receptor Subtypes
Other Angiotensins
Pathophysiology of Renovascular Hypertension
One-Kidney, One-Clip Model
Acute Phase
Renin dependency
Transitional Phase
Chronic Phase
Pathophysiology of Ischemic Nephropathy
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Prosthetic Surgery for Erectile Dysfunction
Prosthetic Surgery for Erectile Dysfunction
 Neuropathic Dysfunction of the Lower Urinary Tract
Neuropathic Dysfunction of the Lower Urinary Tract
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Renovascular Hypertension and Ischemic Nephropathy
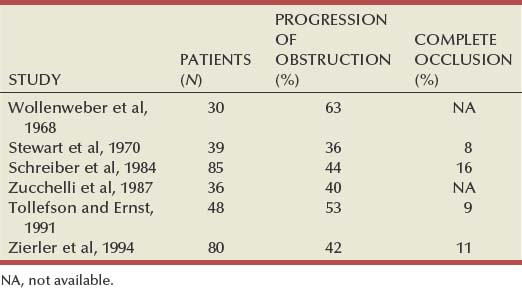
• The finding of anatomical stenosis does not denote arterial hypertension; renal artery stenosis has to be functionally significant to produce renovascular hypertension.