Renal Osteodystrophy: Introduction
The kidneys play a crucial role in the regulation of mineral metabolism by serving both excretory and endocrine functions. Each of these components becomes compromised as renal function declines. In the broadest sense, the term renal osteodystrophy encompasses all of the disorders of bone and mineral metabolism that are associated with chronic kidney disease (CKD). Often, however, the term is used more narrowly to describe the various skeletal disorders and their histologic manifestations among patients with renal dysfunction.
Disturbances in calcium, phosphorus, and vitamin D metabolism, alterations in the regulation of parathyroid hormone (PTH) synthesis and secretion, and factors that lead to the development of parathyroid gland hyperplasia are key components in the pathogenesis of renal bone disease. Additional pathogenic factors include systemic acidosis, aluminum retention, and the accumulation of β2-microglobulin (β2M) in bone and joints. The skeletal manifestations of renal bone disease in individual patients are determined ultimately by the interplay among one or more of these causative factors.
Essentials of Diagnosis
- Disorder of bone and mineral metabolism associated with chronic renal disease.
- Skeletal disorders associated with renal dysfunction.
- Disturbances in calcium, phosphorus, and vitamin D metabolism.
- Parathyroid gland hyperplasia.
- Systemic acidosis, aluminum retention, accumulation of β2M in bone and joints.
Pathogenesis
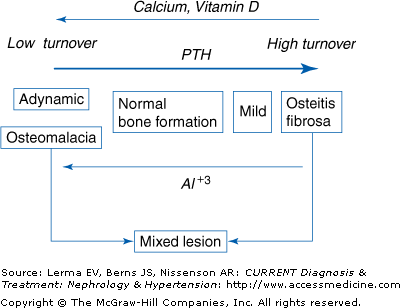
The renal bone diseases represent a spectrum of skeletal disorders ranging from high-turnover lesions arising predominantly from excess PTH secretion to low-turnover lesions of diverse etiology that are typically associated with normal or reduced plasma PTH levels (Figure 20–1). Transitions among histologic subtypes are determined by one or more dominant pathogenic factors. Such changes can be documented by bone biopsy and quantitative bone histology, which represent the definitive method for the diagnosis of renal osteodystrophy. Because plasma PTH levels are a major determinant of bone formation and turnover among patients with CKD, alterations in parathyroid gland function play a key role in the pathogenesis and evolution of renal osteodystrophy. Other factors, however, including diabetes, age-related bone loss, postmenopausal osteoporosis, gender, and race have been recognized increasingly as potentially important additional modifiers of skeletal metabolism and bone turnover among patients undergoing dialysis regularly. Such considerations are particularly relevant given the evolving demographics of the dialysis population in the United States, which is composed increasingly of persons older than 65 years of age and those with diabetes.
Several factors contribute to sustained increases in plasma PTH levels and, ultimately, to the development of high-turnover skeletal lesions in patients with chronic renal failure. Among these are hypocalcemia, impaired renal calcitriol, or 1,25-dihydroxyvitamin D, production, skeletal resistance to the calcemic actions of PTH, alterations in the regulation of prepro-PTH gene transcription, reductions in vitamin D receptor (VDR) and calcium-sensing receptor (CaSR) expression in the parathyroids, and hyperphosphatemia due to diminished renal phosphorus excretion.
Because blood ionized calcium levels represent the most immediate stimulus for PTH secretion, disturbances that lead to hypocalcemia in patients with kidney disease promote excess PTH secretion. Renal 1,25-dihydroxyvitamin D production serves to maintain serum calcium levels by promoting active intestinal calcium absorption, by facilitating calcium release from bone, and by enhancing renal tubular calcium reabsorption. Serum calcitriol levels fall progressively, however, as renal function declines with wide-ranging effects on mineral homeostasis. Although serum 1,25-dihydroxyvitamin D levels vary considerably at any given level of renal function, the proportion of patients with subnormal values increases as renal failure worsens. Such changes account, at least in part, for impaired intestinal calcium absorption and for moderate reductions in serum calcium concentration in many patients with moderate to advanced renal failure. Diminished VDR expression in intestinal epithelial cells may contribute to this disturbance.
Skeletal resistance to the calcemic actions of PTH further compromises the ability to maintain serum calcium levels in those with advanced renal disease. As a result, higher serum PTH levels are required to elicit equivalent biologic responses in patients with chronic renal failure. Abnormalities in vitamin D metabolism and alterations in VDR expression may contribute to this abnormality.
Because calcitriol is a potent inhibitor of cell proliferation, disturbances in renal 1,25-dihydroxyvitamin production and/or reductions in VDR expression have been thought traditionally to contribute to the development of parathyroid hyperplasia in CKD. Vitamin D receptor expression is markedly reduced in parathyroid tissues that exhibit a nodular pattern of tissue hyperplasia, whereas decreased reductions occur in glands with diffuse chief-cell hyperplasia. Moreover, the extent of glandular enlargement is generally greater in the nodular form of parathyroid hyperplasia.
The development and progression of parathyroid gland hyperplasia are a critically important components of renal secondary hyperparathyroidism. Once established, parathyroid enlargement is difficult to reverse because the rate of apoptosis in parathyroid tissue is quite low and the half-life of parathyroid cells has been estimated to exceed 30 years. Clinical assessments of parathyroid gland function demonstrate that differences in functional parathyroid gland size account largely for wide variations in basal plasma PTH levels among patients with advanced renal failure. The secretion of PTH from massively enlarged parathyroid glands may ultimately become uncontrolled due to the ongoing nonsuppressible component of calcium-regulated PTH release thus leading to hypercalcemia and progressive bone disease in patients with advanced CKD.
Phosphorus retention and hyperphosphatemia have been recognized for many years as important factors in the pathogenesis of secondary hyperparathyroidism. The disorder can be prevented in experimental animals with chronic renal failure when dietary phosphorus intake is reduced in proportion to glomerular filtration rate (GFR). Dietary phosphate restriction also lowers plasma PTH levels in patients with moderate renal failure.
Phosphorus retention and hyperphosphatemia appear to aggravate secondary hyperparathyroidism in several ways. Marked elevations in serum phosphorus concentration may lead to the formation of soluble complexes of calcium and phosphorus in plasma, lower blood ionized calcium concentrations, and thus stimulate PTH secretion. Phosphorus abundance impairs renal 1α-hydroxylase activity directly and reduces 1,25-dihydroxyvitamin D synthesis. Either phosphorus retention or renal failure per se may affect posttranscriptional events that influence PTH mRNA stability and hormone synthesis. Finally, phosphorus retention can aggravate parathyroid gland hyperplasia by altering the expression of factors involved in cell cycle regulation and parathyroid cell proliferation.
The skeletal manifestations of hyperparathyroidism are often more pronounced in patients with secondary as compared to those with primary hyperparathyroidism, probably because of the very high plasma PTH levels that occur in renal failure. Values are typically 5- to 10-fold above the upper limit of normal among patients with secondary hyperparathyroidism due to advanced CKD, and they may reach levels that are 20–40 times higher than normal. By contrast, plasma PTH levels in most patients with primary hyperparathyroidism are only 2- to 3-fold above the upper limit of normal.
Unlike those with advanced renal disease who are treated with dialysis, patients with moderate renal insufficiency often have overt histologic evidence of secondary hyperparathyroidism when PTH levels are only modestly elevated. The disparity in disease severity despite markedly different plasma PTH levels between patients with moderate and advanced renal failure is probably attributable to differences in skeletal resistance to the biologic actions of PTH and/or to disturbances in vitamin D metabolism.
Adynamic bone and osteomalacia: In the past, secondary hyperparathyroidism developed almost invariably in untreated patients with progressive renal disease. More recently, however, fewer patients have markedly elevated plasma PTH levels when regular dialysis is begun. Many have bone biopsy evidence of adynamic renal osteodystrophy, which is characterized by subnormal rates of bone formation and turnover. As mentioned previously, changes in the demographics of the dialysis population may account for this change.
Adynamic renal osteodystrophy currently accounts for most cases of low-turnover bone disease in patients undergoing dialysis regularly. Osteomalacia is seen much less often. Approximately 40% of those treated with hemodialysis and more than 50% of those receiving peritoneal dialysis have plasma PTH levels that are only modestly elevated or that fall within the normal reference range. Such values are typically associated with reduced rates of bone formation and turnover without evidence of defective skeletal mineralization.
In the 1970s and 1980s, aluminum retention and bone aluminum accumulation accounted for most cases of adynamic bone and osteomalacia among patients with CKD. Two distinct patterns of aluminum exposure were described. One was due to inadequate water purification during the preparation of dialysis solutions, which led to inadvertent parenteral aluminum loading. The other was due to the long-term ingestion of aluminum-containing, phosphate-binding agents, which led to gradual aluminum loading from intestinal aluminum absorption. Bone aluminum deposition was a prominent finding both in patients with adynamic bone and in those with osteomalacia. Bone and muscle pain, proximal myopathy, and skeletal fracture were common. Bone histology improved and bone formation increased when aluminum overload was treated effectively.
Aluminum has diverse effects on bone and mineral metabolism. It inhibits the proliferation and the differentiated function of osteoblasts, reduces collagen synthesis, and suppresses PTH secretion. Accordingly, adynamic bone due to aluminum retention may arise both through direct inhibitory actions of aluminum on osteoblasts and through indirect effects on bone cell activity that are mediated by reductions in parathyroid gland function and low plasma PTH levels. Aluminum also interferes directly with skeletal mineralization to cause osteomalacia.
Risk factors for aluminum-related bone disease include previous parathyroidectomy, a history of renal transplantation and graft failure, bilateral nephrectomy, and diabetes mellitus. By promoting the formation of soluble complexes with aluminum, citrate markedly enhances intestinal aluminum absorption, and the use of citrate-containing compounds in patients with renal failure who are also ingesting aluminum-containing medications should be avoided. High plasma PTH levels appear to partially offset the adverse skeletal effects of aluminum. This may account for the somewhat greater risk of aluminum-related bone disease among diabetic patients and those who have undergone parathyroidectomy previously, conditions associated with low plasma PTH levels.
Fortunately, aluminum-related bone disease is now uncommon. Diabetes, corticosteroid therapy, and increasing age account for adynamic lesions in many patients. In this regard, the proportion of diabetic and elderly patients in the dialysis population continues to increase. Many of the histologic features of adynamic renal osteodystrophy are indistinguishable from those of osteoporosis from any of a variety of causes. The possibility of osteoporosis from causes other than renal bone disease must be considered when reductions in trabecular bone volume or cortical thinning are prominent histologic features in patients with CKD because such changes are not integral components of adynamic renal osteodystrophy.
The widespread use of large doses of oral calcium as a phosphate-binding agent and the use of large doses of active vitamin D sterols to treat secondary hyperparathyroidism may account for the increased prevalence of adynamic bone among patients receiving dialysis. Both interventions can lead to sustained reductions in plasma PTH levels. Calcitriol may also diminish osteoblastic activity directly when given in large intermittent doses to patients undergoing regular dialysis.
The long-term consequences of adynamic renal osteodystrophy, when not due to aluminum toxicity, remain uncertain. Some studies suggest that the risk of skeletal fracture may be greater among patients with relatively low plasma PTH levels, although the data are not conclusive. The prevalence and extent of arterial calcification have been reported to be greater among adult dialysis patients with adynamic skeletal lesions than in those with hyperparathyroidism. Episodes of hypercalcemia, which occur more often in those with adynamic renal osteodystrophy, may be contributory. In prepubertal children, adynamic skeletal lesions have been reported to be associated with reductions in linear growth.
For patients with osteomalacia, aluminum toxicity must be excluded if there is a history of sustained aluminum ingestion or if there are concerns about the adequacy of water purification procedures in dialysis facilities. Evidence of inadequate vitamin D nutrition, which is common among patients with CKD, should be sought by measuring serum 25-hydroxyvitamin D levels, and vitamin D nutrition should be restored by the administration of cholecalciferol or ergocalciferol if values are below 30 ng/mL. Long-term treatment with phenytoin and/or phenobarbital can lead to osteomalacia in nonuremic persons, and a higher prevalence of symptomatic bone disease has been reported in dialysis patients receiving these drugs. Persistent hypocalcemia and/or hypophosphatemia can lead to osteomalacia in some patients, but the disorder is seen much less frequently now that aluminum-containing medications are used sparingly and more attention has been given to maintaining adequate calcium and vitamin D nutrition among patients with CKD.
Some patients with CKD have histologic features of both osteitis fibrosa and osteomalacia, a disorder known as the mixed lesion of renal osteodystrophy. There is biochemical evidence of secondary hyperparathyroidism, but other factors account for defects in skeletal mineralization. Persistent hypocalcemia and/or hypophosphatemia are found in some patients, and nutritional vitamin D deficiency is present in others. Mixed lesions of renal osteodystrophy can be seen in patients with osteitis fibrosa who are in the process of developing aluminum-related bone disease or in those with aluminum-related osteomalacia who respond favorably to treatment with deferoxamine (DFO) with increases in bone formation. Mixed renal osteodystrophy can thus represent a transitional state between the high-turnover lesions of secondary hyperparathyroidism and the low-turnover disorders of osteomalacia or adynamic bone.
Clinical Findings
The signs and symptoms of renal bone disease are rather nonspecific. Both the extent of various laboratory abnormalities and the severity of certain radiographic changes often fail to correspond to the clinical manifestations. Many patients report few symptoms despite striking disturbances in biochemical and x-ray parameters, but subtle complaints of musculoskeletal discomfort are common when sought by specific inquiry. Bone pain and muscle weakness are common. Skeletal deformities can develop in advanced cases, and extraskeletal calcifications are frequent.
Bone pain is often present in patients with renal osteodystrophy. The onset is insidious with symptoms progressing gradually over many months or years. Pain is diffuse and nonspecific, and it is often aggravated by bearing weight and by changes in posture. When localized, the lower back, hips, and legs are affected most often. Pain in the heels or ankles may be a prominent complaint. Some patients experience an acute arthritis or periarthritis that is unrelieved by massage or the application of heat locally. Severe bone pain is more common in patients with aluminum related bone disease than in those with osteitis fibrosa, and it is a prominent clinical feature of bone aluminum toxicity. Nevertheless, some patients with advanced secondary hyperparathyroidism are incapacitated substantially. Physical examination is generally unremarkable unless skeletal fractures have occurred or unless bony deformities have developed.
Proximal myopathy occurs in some patients with advanced renal failure. Symptoms appear slowly, and weakness and aching of the muscles are the most common manifestations. The physiologic basis of this disorder is not understood. Favorable clinical responses have been noted in some patients after treatment with calcitriol or 25-hydroxyvitamin D, following parathyroidectomy, after successful renal transplantation, or during treatment of aluminum-related bone disease with DFO. The role of abnormal vitamin D metabolism in the pathogenesis of uremic myopathy remains uncertain, but a careful evaluation must be done to exclude severe secondary hyperparathyroidism or bone aluminum toxicity. An empirical therapeutic trial of calcitriol or 25-hydroxyvitamin D is warranted in patients with persistent complaints of muscle pain and weakness.
In patients with aluminum-related bone disease, skeletal deformities are confined predominantly to the axial skeleton and include lumbar scoliosis, kyphosis, and distortion of the thoracic cage. Some persons with severe osteitis fibrosa develop rib deformities and pseudoclubbing.









