Lane S. Palmer, MD, FAAP, FACS, Howard Trachtman, MD The glomerular filtration rate (GFR) at birth is 10% to 15% of the normal adult value. The decrease in function is caused by three factors: lower intraglomerular perfusion pressure, decreased ultrafiltration coefficient of the filtration barrier, and diminished filtration surface area. Of these three factors the final one is quantitatively the most important and is due to a combination of decreased number and perfusion of the cortical nephrons (Spitzer, 2003). Increased circulating levels of vasoactive hormones such as vasopressin and plasma renin activity and reduced synthesis of vasodilatory factors such as nitric oxide mediate the reduced glomerular blood flow. The reduction in filtration surface is exacerbated in very low–birth-weight infants delivered at 25 to 28 weeks’ gestation because the premature delivery curtails the normal process of nephron development and glomerulogenesis (Ichikawa et al, 2002). In utero obstruction interferes with normal glomerular development and triggers tubular atrophy and interstitial fibrosis. This is associated with release of profibrotic cytokines such as transforming growth factor-β, macrophage infiltration, and epithelial-to-mesenchymal transformation (Bascands and Schanstra, 2005; Grande and Lopez-Novoa, 2009). Using experimental murine models, in which unilateral ureteral obstruction is created before completion of nephrogenesis, reversal of the obstruction leads to an increased tubular repair, reduced macrophage number, and less collagen accumulation (Cochrane et al, 2005). This suggests that correction of obstructive lesions prenatally could restore normal renal growth and preserve renal function. Renal sodium handling is impaired in the fetus and infant. This is reflected in a higher fractional excretion of sodium (FENa) in low-birth-weight infants and even full-term neonates. In part this reflects increased permeability of the proximal and distal tubule. In addition, the response of the distal nephron to antinatriuretic hormones such as aldosterone is impaired. The urinary concentrating capacity is diminished in fetal life and in infants owing to reduced GFR and delivery of filtrate to the diluting segment of the nephron, shorter loops of Henle, and limited response of the collecting tubule to vasopressin. The renal acidification is limited in neonates because of lower sodium-hydrogen exchange in the proximal tubule, decreased bicarbonate reabsorption in the proximal tubule secondary to relative expansion of the extracellular fluid compartment, and reduced expression and activity of the Na+,K+-ATPase pump that drives hydrogen ion secretion. The alterations in sodium and hydrogen balance result in compromised potassium secretion into the final urine. The potassium retention in neonates is due to an absence of apical secretory potassium channels in the distal tubule and a predominance of the H+,K+-ATPase in the apical membrane that mediates potassium reabsorption (Gurkan et al, 2007). The inability to excrete potassium is also the consequence of reduced number of renal outer medullary potassium (ROMK) channels and diminished potassium recycling in the loop of Henle. In addition to the short-term impact of premature delivery and low birth weight, these two factors may have important long-term ramifications for kidney health. Beginning in the mid 1980s, reports appeared in the literature indicating that low-birth-weight infants were at higher risk of developing hypertension and secondary renal diseases such as diabetic nephropathy in adulthood compared with infants of normal weight. These epidemiologic linkages were strengthened by the detection of a lower number of nephrons in adults with hypertension who died in early adulthood after sudden unexpected accidents. The adverse outcomes have been attributed to the long-term consequences of being born with a diminished number of nephrons—sustained hyperfiltration and glomerular hypertrophy (Barker et al, 2006). The situation is analogous to the case of infants born with a solitary kidney who can develop proteinuria and focal segmental glomerulosclerosis (FSGS) at a relatively young age. The long-term implications of reduced nephron number at birth continue to be a subject of ongoing study (Vikse et al, 2008) and may represent a combined effect of both low birth weight and prematurity. There is uncertainty whether the consequences of low nephron number apply to both boys and girls and to all racial and ethnic groups. Finally, recent data suggest that impaired postnatal growth may exacerbate the maladaptive response to low birth weight (Bacchetta et al, 2009). Key Points Transition from Fetus to Infant The composition of the two major body water compartments—ECF and intracellular fluid (ICF)—varies greatly. In the ECF the principal cation is sodium and the major anions are chloride and bicarbonate. In the ICF, in contrast, potassium is the major cation and proteins and phosphate comprise the bulk of the anions. However, in both compartments there is electroneutrality and an equal number of osmotically active solutes. The major factor leading to the differential distribution of solutes between the ECF and ICF is the Na+,K+-ATPase pump, which actively extrudes sodium from the cells. Because sodium is confined to the ECF, disturbances in the ECF volume reflect abnormalities in external sodium balance. Thus, if intake of sodium exceeds output, then the patient will have ECF volume expansion with clinical symptoms such as hypertension, pulmonary edema, and peripheral edema. If sodium losses are not counterbalanced by sufficient dietary intake, then the patient will develop intravascular volume contraction, manifesting as tachycardia, hypotension, and impaired peripheral perfusion. Serum osmolality is the primary determinant of the normal distribution of water between the ECF and ICF in a 1 : 2 ratio. Hyperosmolality causes ICF contraction, and hypo-osmolality causes ICF expansion. The major clinical consequences of changes in the ECF : ICF ratio occur because of altered cerebral volume and brain dysfunction, leading to irritability, lethargy, somnolence, and seizure (Trachtman, 1991). Intravenous fluid therapy should be considered a form of drug treatment and should be instituted only if there are appropriate clinical indications. These include inability to tolerate enteral fluids, severe ongoing losses, medical need to avoid oral intake, and acute kidney injury in which close monitoring of fluid intake may be necessary. Under these circumstances, maintenance fluids are provided to supply sufficient amounts of water, sodium, and potassium to account for normal urinary and gastrointestinal losses and insensible losses through the skin and respiratory tract. These quantities are best calculated based on surface area, although calculations based on body weight, a more accessible measurement, are generally reliable. Maintenance fluids need to be adjusted upward in children with fever, tachypnea, or dermatologic diseases that disrupt the skin barrier. For example, premature infants have increased insensible losses because of their thin skin and require increased free water intake to prevent hypernatremia. The maintenance fluid requirements are generally provided as a hypotonic solution of sodium chloride containing approximately 50 mmol/L. Recently, controversy has arisen about the appropriate composition of parenteral maintenance fluids. There are those who advocate routine administration of isotonic saline (0.9% or NaCl 154 mmol/L) to prevent hospital-acquired hyponatremia (Moritz and Ayus, 2003). Others suggest that the standard fluid should continue to be a hypotonic solution with recognition that modification may be necessary in seriously ill children who may be at risk of altered vasopressin release and hyponatremia (Holliday et al, 2004). Absent clinical trials, clinicians are advised to be aware of the risks of parenteral therapy in hospitalized children and to monitor the effects of prescribed fluid treatment. Children with severe intravascular volume contraction should receive rapid infusion of isotonic saline to stabilize cardiovascular status, regardless of the serum sodium concentration. In the majority, in which there is isotonic dehydration, the deficit can be repaired steadily over 24 hours. In patients with hyponatremia or hypernatremia the fluid deficit should be reversed more slowly over at least 48 hours to prevent adverse consequences of cerebral cell swelling in patients with hypernatremia or cerebral cell shrinkage in patients with hyponatremia. In children with hypernatremia it is important to infuse hypotonic fluids but at a slow rate to enable gradual normalization of hyperosmolality (Banister et al, 1975). Children with nephrogenic diabetes (see later) are prone to excessive free water loss and hypernatremia (Adrogue and Madias, 2000). Similarly, patients with obstructive uropathy and pseudohypoaldosteronism are prone to develop hyponatremia, especially if they have a concurrent urinary tract infection. These disturbances need to be taken into account when formulating fluid regimens. Isolated hematuria is a very common problem, the treatment of which is often shared by nephrologists and urologists. Studies done in a wide range of countries around the world indicate that the frequency of detecting blood in the urine in pediatric patients ranges from 0.5% to 3% (Hogg, 2009). Most of the time the problem is self-limited, resolves spontaneously, and is not a sign of underlying disease. Thus although mass screening of urine samples in pediatric patients is common in several Asian countries, there is a movement away from this practice. In 2007 the American Academy of Pediatrics revised its clinical recommendations and no longer suggests performing a urinalysis routinely at any age during childhood. The majority of cases of hematuria are microscopic. The differential diagnosis (Table 112–1) in patients with gross hematuria is no different than in those with microscopic hematuria (Youn et al, 2006; Greenfield et al, 2007). The key difference is that patients with gross hematuria are more likely to be referred for urgent assessment and the likelihood of finding an underlying cause. In studies of children with gross hematuria, nearly 60% have nonglomerular causes, 25% have a glomerular etiology, and the remainder have no established cause. Table 112–1 Causes of Hematuria in Children Studies of hematuria in children have been conducted by screening healthy children or by retrospective analyses of children with hematuria. Vehaskari (1979) estimated the prevalence of hematuria in children to be 1.1% (excluding those with transient hematuria): 74% of the 8954 children had hematuria in only one sample, 83 of the remaining 131 children with persistent hematuria had isolated hematuria most commonly due to urinary tract infection (7%), whereas 59 had associated proteinuria; this study was conducted before hypercalciuria was identified as a common cause of hematuria. Hisano (1991) found 251 of 160,000 screened junior high school students had isolated microscopic hematuria with an identifiable cause in 115: menses (35%), urinary tract infection (5.6%), hypercalciuria (2%), Henoch-Schönlein purpura (1.2%), and hydronephrosis (1.2%). Approximately 25% of children had resolution of hematuria, less than 1% had proteinuria, and none developed renal insufficiency. Turi (1989) reported a retrospective analysis of 341 children, ages 2 to 12 years with persistent, isolated hematuria of at least 6 months’ duration followed for 2 to 17 years. Hematuria was isolated microscopic in 31%, macroscopic in 28%, or both in 41%. Persistent hematuria was documented monthly in 40%. Renal biopsy in 47 children for development of a nephritic urine sediment or proteinuria demonstrated mesangial proliferative glomerulonephritis and hereditary nephritis most commonly. Twenty percent of subjects had hypercalciuria and/or nephrolithiasis. The remaining 226 children had isolated hematuria, which resolved in 163 children. These studies support the concept that isolated asymptomatic hematuria is a relatively benign condition after exclusion of children with easily identifiable causes by history and physical examination, family history, urine microscopy, renal imaging, urine culture, urinary calcium excretion, and renal biopsy, if indicated. Persistent microscopic hematuria lasting at least 6 months or gross hematuria warrants investigation. The evaluation of hematuria includes a proper history, physical examination, and laboratory and radiographic studies (Fig. 112–1). The history should include questions regarding the timing of the presence of the hematuria as an isolated or recurrent event; hematuria that occurs throughout voiding is more likely to be glomerular, whereas terminal hematuria is more likely to be nonglomerular. Benign urethrorrhagia presents as postvoid droplets of blood from the meatus after a grossly clear void (Kaplan and Brock, 1982). Glomerular hematuria is often described as urine that is tea or cola colored, whereas urine of nonglomerular hematuria is more likely to be red. Certain diagnoses may be suspected based on the presence of associated symptoms such as dysuria, urgency, frequency, abdominal pain, prior respiratory infection, lower extremity swelling, or symptoms of hypertension (headaches, blurred vision), joint pain, fevers, weight loss, rash, or antecedent streptococcal infection. A patient or family history of sickle cell disease or trait, deafness, hematuria, or renal disease should be sought. The physical examination should assess for blood pressure with a proper-sized sphygmomanometer cuff, abdominal tenderness or masses, costovertebral angle tenderness, ecchymosis, rashes, periorbital or lower extremity swelling, and signs of trauma on the flank, pelvis, or genitalia. A fresh urine sample should be collected for analysis and culture. Hematuria is defined as the presence of five or more red blood cells per high-power microscope field (Fitzwater and Wyatt, 1994). Hematuria is detected by dipstick via a reaction of a peroxidase with hemoglobin; however, the dipstick test may be falsely positive in the presence of hemoglobinuria or myoglobinuria. Fresh-spun urine should also be screened for protein, glucose, nitrates, leukoesterase, bilirubin, specific gravity, and pH. The key diagnostic test is a microscopic examination of the urine sediment. Patients with glomerular hematuria will have red blood cell casts and dysmorphic erythrocytes (irregular shape and size with distorted contours) in their sample, whereas children with nonglomerular hematuria have eumorphic red erythrocytes (uniform in size resembling circulating red blood cells) without red blood cell casts. This distinction can usually be made reliably by examining the urine directly (Trachtman, 2006). A 2- to 4-hour delay in urine inspection can cloud the distinction between nonglomerular and glomerular bleeding. If prompt examination of freshly voided urine samples is not feasible in a specific clinic setting, the addition of Cellfix, a formaldehyde-based fixative, allows preservation of red cell morphology for at least 24 hours and can enhance the diagnostic value of the microscopy portion of the urinalysis (Komarova et al, 2003). Phase contrast microscopy has been touted to enhance accuracy. In addition, some clinicians have recommended measurement of red cell size with Coulter counter devices used in hematology clinics. Patients with dysmorphic red cells in the urine will have smaller erythrocyte volume and greater scatter in cell size compared with patients with nonglomerular hematuria. Microscopic evaluation of the specimen should be performed to qualify and quantify the extent of the hematuria and the presence of crystals (uric acid, cystine) and casts (red or white cell). A renal and bladder ultrasound evaluation will screen for possible renal parenchymal or bladder causes of the hematuria. Computed tomography best defines renal masses, stones, and nutcracker syndrome, in which there is extrinsic compression on the aberrantly located renal vein. Additional information may be gleaned by magnetic resonance imaging (MRI). These modalities would be used to better characterize any abnormalities identified by ultrasonography. Voiding cystourethography is reserved for children with culture-positive infection or to further evaluate any abnormal ultrasound findings in the bladder. There is no role for cystoscopy in the evaluation of microscopic hematuria (Feld et al, 1998) if the sonogram is normal. Small tumors may be missed but are benign (Kaplan, 1985). Cystoscopy for gross hematuria can be useful if bleeding occurs at the time of the study. If bloody effluent is observed from both ureteral orifices, a glomerular source is more likely. If bloody urine is observed from only one ureteral orifice, unilateral urinary tract or vascular disease may be present and retrograde pyelography or ureteroscopy may be indicated. Hypercalciuria can present as isolated microscopic hematuria, dysuria, or recurrent gross hematuria. Gross hematuria secondary to hypercalciuria can be distinguished from that due to glomerular diseases by the potential presence of dysuria, clots in the urine, and rapid resolution of the hematuria within 24 hours. It should be noted that hypercalciuria can also cause dysuria and mimic a urinary tract infection in which the urine culture shows no bacterial growth. Finally, hypercalciuria can be the major factor leading to urolithiasis. Numerous dietary factors such as increased intake of sodium and magnesium influence tubular reabsorption of calcium and can cause hypercalciuria (Bonny et al, 2008). Hypercalciuria may have a genetic component, accounting for the clustering of kidney stones in family pedigrees. For example, specific polymorphisms in the calcium-sensing receptor gene are associated with a gain-of-function and increased susceptibility to idiopathic hypercalciuria (Vezzoli et al, 2007). The diagnosis of hypercalciuria is best made based on a 24-hour urine collection in which the calcium excretion exceeds 4 mg/kg/day. Because of logistic issues involved in collecting 24-hour collections, clinicians often rely on the calcium-to-creatinine ratio in a spot sample. The normal calcium-to-creatinine ratio value declines with age, and in children older than 4 years old the ratio should be less than 0.2. There are a number of secondary causes of hypercalciuria, including RTA, hyperparathyroidism, malignancies, immobilization, and sarcoidosis. However, in most cases it is idiopathic. In the past, effort was directed at discriminating between hyperabsorption of calcium from the gut versus renal leak of calcium as the basis for hypercalciuria. However, this distinction is no longer considered clinically relevant because it does not influence the diagnostic workup or patient management. In addition to these renal manifestations, idiopathic hypercalciuria predicts bone loss. Studies have demonstrated an association between the degree of hypercalciuria and bone density of the femoral neck in adult men and women (Asplin et al, 2006). The long-term implications of this finding require ongoing study. Treatment of hypercalciuria by increasing fluid intake should be started on all diagnosed children, with thiazide administration reserved for symptomatic children unresponsive to conservative measures or those with high urinary levels or active stone disease. The filtration mechanism of the glomerulus aims to limit the passage of macromolecules, such as protein, from the blood into the urine. The pores in the slit diaphragm between the foot processes have a 40-Å diameter, which allows passage of low-molecular-weight molecules (α1 and β1 microglobulins) that are reabsorbed by the proximal tubule and therefore not detected in the urine in the absence of tubular damage (Tomlinson, 1992). The negative charge of the glomerular capillary wall provided by heparin sulfate and proteoglycans repel other negatively charged molecules, such as albumin. The loss of the negative charge or breakdown of the size barrier can lead to proteinuria. A third factor involved in filtration is the presence of hydrostatic and oncotic forces as blood traverses the glomerulus. Proteinuria can arise from hyperfiltration when a reduction of functioning nephrons occurs (e.g., from nephrectomy). Recent studies have created controversy about the pathophysiologic basis of glomerular proteinuria—excessive leak due to a primary disturbance in glomerular barrier function or failure to reabsorb the filtered load in the proximal tubule (Russo et al, 2007). Abnormal urinary protein excretion is defined as greater than 4 mg/m2/hr (Nephrotic syndrome in children…, 1979) in an overnight urine collection or a protein-to-creatinine (mg/mg) ratio greater than 0.2 in a first morning urine sample. Proteinuria is prevalent in 5% to 15% of children (McKenzie et al, 1964) and is associated with increasing age with a peak incidence of 13 years in girls and 16 years in boys (Wagner et al, 1968). It is common to find isolated proteinuria, but persistent proteinuria indicates renal disease and warrants evaluation (Vehaskari and Rapola, 1982). Proteinuria is important because it may reflect progressive renal disease (Kannel et al, 1984; Williams and Coles, 1994) and lead to hypocholesterolemia and hypertriglyceridemia, which pose a higher cardiovascular risk (Sytkowski et al, 1990). Similarly, medications aimed at reducing proteinuria can delay progressive renal failure (Remuzzi et al, 2004). Proteinuria is first detected by urinary dipstick testing, which primarily detects albumin (Fig. 112–2). The colorimetric reaction between albumin and tetrabromophenol blue results in variable shades of green, depending on the approximate concentration of protein in the urine: trace indicates approximately 15 mg/dL; 1+, 30 mg/dL; 2+, 100 mg/dL; 3+, 300 mg/dL; and 4+, 2000 mg/dL or more. False-positive urine protein levels may be caused by alkaline urine, antiseptic cleanser, radiocontrast agents, or a highly concentrated sample. False-negative urine protein levels may be found in very dilute urine. Significant proteinuria is indicated by a urine sample with a dipstick reading of 1+ and a specific gravity of at least 1.015. Confirmation of the level of proteinuria may be performed using sulfosalicylic acid, which uses protein precipitation and turbidity to estimate the level of proteinuria (Hogg et al, 2000). In the absence of other signs or symptoms of renal or urinary tract disease, including systemic disease, the urinalysis may be repeated in several weeks on the first morning specimen to confirm proteinuria. Quantification of the proteinuria is best performed from a 24-hour urine collection. Normal urine protein excretion in children is less than 4 mg/m2/hr (<100 mg/m2/24 hr). Protein excretion rate greater than 4 mg/m2/hr is abnormal, and greater than 40 mg/m2/hr is nephrotic-range proteinuria. Urinary creatinine value should be measured (normal is 15 to 20 mg/kg/day in girls and 20 to 25 mg/kg/day for boys) to confirm the adequacy of the collection. However, timed urine collection can be difficult to accomplish and therefore inaccurate, particularly in the incontinent child (Hogg et al, 2000). Moreover, it is not useful in establishing the diagnosis of orthostatic proteinuria, a variant of normal. A random urine protein-to-creatinine ratio correlates well with 24-hour quantification. The normal values are less than 0.5 for children 6 to 24 months of age and less than 0.2 for children older than 2 years; a ratio above 3.5 is considered in the nephrotic range. In a first morning sample, protein-to-creatinine values greater than 2.0 indicate nephrotic range proteinuria. Significant proteinuria can occur secondary to glomerular or tubulointerstitial diseases, but it is generally due to glomerulopathies. Proteinuria may be categorized as transient, orthostatic, or fixed/persistent. Transient proteinuria is present in 30% to 50% of children with proteinuria and may be associated with fever, stress, dehydration, and exercise. Transient proteinuria associated with a fever rarely exceeds 2+ on the dipstick and abates with the fever. Exercise-induced proteinuria increases with age (Poortmans et al, 1996), is related to the vigor of exercise and not duration, (Poortmans, 1985), rarely exceeds 2+ on dipstick, and resolves within 48 hours of rest. Postural (or orthostatic) proteinuria is common (60% of asymptomatic children) (Norman, 1987) in which protein excretion is normal while supine but becomes abnormal when erect. Children are asymptomatic, and the only abnormal laboratory finding is an elevated urine protein level after being upright. Postural proteinuria may be documented by collecting two timed urine samples and finding no proteinuria while the child is recumbent (sleeping) and proteinuria while the child is upright. The 24-hour urinary protein excretion in children with orthostatic proteinuria should not exceed 1000 mg/day. Persistent proteinuria (up to 6% of school-aged children (Vehaskari and Rapola, 1982; Yoshikawa et al, 1991) is diagnosed after confirming proteinuria on two dipstick tests with at least 1+ at least 1 week apart and excluding postural proteinuria. Its etiology can be divided as either glomerular where there is increased permeability of the glomerulus or as tubular from decreased protein reabsorption. The glomerular type is more common and can be selective (molecules smaller than albumin) or nonselective (molecules larger than albumin). The differential diagnosis for isolated proteinuria includes rare cases of minimal change nephrotic syndrome, FSGS, membranous nephropathy, or IgA nephropathy. Although FSGS is more frequent in African-American children, IgA nephropathy is much more common in white children (Nair and Walker, 2006). Membranous nephropathy is a rare disease across the entire pediatric age range (Chen et al, 2007). Obstructive uropathy is thought to be a common cause of tubular proteinuria, but it usually presents as other signs and symptoms. The measurement of microalbuminuria is generally confined to the management of diabetic nephropathy. Tests to measure selectivity of proteinuria or to quantitate low-molecular-weight proteins are not routinely available and of uncertain value. Unlike the situation in adult patients, the long-term implications of microalbuminuria on general health and the incidence of cardiovascular disease is unknown in pediatric patients. In fact, because of the high prevalence of orthostatic proteinuria in adolescents, testing the early morning sample is both more appropriate and more feasible than measuring proteinuria in a 24-hour collection. The diagnosis of the cause of proteinuria usually requires performance of a kidney biopsy. In 70% of children with isolated proteinuria, the urinary abnormality spontaneously resolves within 12 months (Trachtman et al, 1994). Therefore criteria for performing a kidney biopsy are (1) persistent subnephrotic proteinuria for at least 12 months or (2) nephrotic-range proteinuria that persists for 3 to 4 months or rising levels of proteinuria in patients who are being followed after the initial detection of isolated proteinuria. However, these standards are likely to vary from center to center reflecting the pattern of disease regional and at each institution. In general, renal biopsies done under ultrasonographic guidance are a safe and well-tolerated procedure (Gauthier et al, 1993). The presence of both hematuria and proteinuria suggests the need for a more comprehensive and urgent evaluation. Nephrotic syndrome is defined as edema, hypoalbuminemia, hyperlipidemia, and heavy proteinuria. The amount of proteinuria considered to be nephrotic is an excretion greater than 40 mg/m2/hr or a random urine protein-to-creatinine ratio of greater than 2.0 to 3.0 (Hogg et al, 2000). Nephrotic syndrome may be subdivided into congenital, primary (from any type of glomerulonephritis), or secondary (systemic disease, malignancy, allergic reaction or nephrotoxin). The incidence of primary nephrotic syndrome is 2 to 4 cases per 100,000 children younger than 19 years old and is most commonly due to minimal change disease (65% to 80%). In general, nephrotic syndrome is a disease of the glomerular podocyte, with the identification of genetic disorders of podocyte proteins providing the basis for this understanding. Hypoalbuminemia occurs from the urinary loss of albumin causing a reduced plasma colloid oncotic pressure and a fluid shift from the intravascular to the interstitial space. This translates clinically into the development of edema as the serum albumin level falls below 2.5 g/dL. The rapid intravascular volume loss leads to compensatory reabsorption of sodium and water by the kidney, which aggravates the edema. In addition to underfilling of the intravascular space, nephrotic edema occurs because of primary sodium retention and overfilling of the circulation (Schrier and Fassett, 1998). While albumin synthesis increases, it is insufficient to replace the ongoing albumin losses, poor dietary protein intake, and increased catabolism of albumin. All infants with primary nephrotic syndrome have proteinuria, hypoalbuminemia, hyperlipidemia, hypogammaglobulinemia, and usually normal renal function. Edema is present at birth or develops over several weeks in concert with increasing proteinuria. Hematuria is uncommon. The leading causes of nephrotic syndrome in infants include congenital nephrotic syndrome–Finnish type (CNF), diffuse mesangial sclerosis (DMS), minimal change nephrotic syndrome (MCNS), and focal sclerosis glomerulonephritis (FSGS). Family history, outcome, and corticosteroid response may be the only ways to secure the diagnosis. MCNS is rare in early infancy, responds to corticosteroids, does not progress to end-state renal disease, shows minimal changes on biopsy, and has a negative family history. FSGS is rare and may respond to corticosteroids. The rarity of MCNS and FSGS and the increased risk of severe infection in infants with CNF preclude starting corticosteroids in the first 3 months of age in the absence of a definitive diagnosis. DMS is rare and clinically indistinguishable from CNF occurring in siblings, is resistant to corticosteroids, and progresses to end-stage renal disease (ESRD) before age 3 years. Early in the disease DMS can be distinguished from CNF on biopsy by its mesangial matrix expansion without hypercellularity, thickened glomerular basement membrane (GBM), crowding of the capillary lumens, and eventual glomerulosclerosis, leaving a contracted sclerotic mass in a dilated Bowman space. DMS is the lesion associated with the glomerulopathy of Denys-Drash syndrome and may occur secondary to mutations in the Wilms tumor-1 (WT1) gene (Chiang et al, 2008). Once the presumptive diagnosis is made, high-dose (60 mg/m2/day) daily treatment with oral prednisone is started (Mendoza and Tune, 1992). This regimen is continued for at least 4 weeks, followed by an additional 4 weeks of 40 mg/m2/day on alternate days and then a gradual taper using alternate-day dosing. To simplify management, the daily dose can be 2 mg/kg and the alternate day dose 1.5 mg/kg (Gipson et al, 2009). Most children respond with remission of proteinuria within 4 weeks (negative or trace protein on dipstick for 3 consecutive days). Dietary salt restriction is another important component of treatment. Approximately two thirds of children will experience relapse, which is treated with high daily doses of corticosteroids until remission and then the dosage is tapered. Cytotoxic agents (cyclophosphamide or chlorambucil) or cyclosporine are used in some children if relapses are frequent or unsustainable without corticosteroids. In cases in which renal biopsy is performed, light microscopy demonstrates normal renal tissue or demonstrates a mild, focal increase in mesangial cellularity and matrix. On electron microscopy, effacement of epithelial cell foot processes, normal glomerular basement membrane thickness, and small paramesangial electron-dense deposits can be seen. These lesions fail to stain or stain slightly positive for IgM. Focal segmental glomerulosclerosis (FSGS) is the second most common cause of primary nephrotic syndrome in children (Bonilla-Felix et al, 1999), accounting for 10% to 20% of cases. Its incidence may be increasing in all ethnic groups, perhaps related to the epidemic of obesity and the metabolic syndrome. FSGS is more common among older children and African-American children and is much less likely to respond to corticosteroid therapy. FSGS has many causes but no clear pathophysiology. Whereas 15% to 25% of children with primary nephrotic syndrome have biopsy-proven FSGS, 20% of patients with FSGS have asymptomatic proteinuria. Microscopic hematuria is present in over 50% of affected children, but gross hematuria is rare. Renal tubular dysfunction (glucosuria, aminoaciduria, RTA, Fanconi syndrome) and concentrating defects are risk factors for progression to chronic renal failure. Children with resolving postinfectious glomerulonephritis will have had an upper respiratory infection with or without confirmed pharyngitis in the recent past, and this is the most common cause of acute glomerulonephritis in children. There is a latency period of 7 to 21 days after a group A β-hemolytic streptococcal infection and the acute phase of glomerulonephritis. At the time of presentation with cola-colored urine the children are well without hypertension, edema, or proteinuria. In these children, confirmation of the diagnosis is contingent on resolution of the hematuria, which may take up to 2 years after the acute episode. The diagnostic studies include a positive antistreptolysin O or streptozyme titer and a decreased serum complement (C3) concentration. However, there is an ongoing paradigm shift in obtaining serologic studies in suspected cases in that there may be no value in routinely measuring the C3 levels because the temporary fall in level during the acute phase of the illness has generally passed and an isolated elevation in antibody titers to streptococcal-related antigens has no diagnostic value (Srisawat et al, 2006). Provided the hematuria gradually resolves, it is thought that the long-term prognosis of the patients is no different than that of healthy controls (Blyth et al, 2007). Complete recovery of renal function can be expected in more than 90% of cases, even in those who have a protracted clinical course requiring dialysis. IgA nephropathy (Berger disease) is more prevalent in the second decade of life and is more common in boys compared with girls. It is usually heralded by recurrent episodes of painless gross hematuria without clots synchronous with upper respiratory tract infections (Davin and Weening, 1999). The gross hematuria takes 3 to 4 days to gradually return to normal; however, some children and adolescents have persistent isolated microscopic hematuria as the sole manifestation of IgA nephropathy. It is important to distinguish IgA nephropathy in which systemic manifestations are uncommon from Henoch-Schönlein purpura, which also can cause nephritis but, unlike IgA nephropathy, be associated with distinctive urologic problems such as acute scrotal swelling (Ha and Lee, 2007). Recent studies have suggested that IgA nephropathy may be the result of abnormal galactosylation of IgA1 molecules (Buck et al, 2008). This triggers the synthesis of autoantibodies, formation of immune complexes, and deposition in the glomerular mesangium (Suzuki et al, 2009). Serology will demonstrate elevated antistreptococcal titers and normal C3 levels. The decision on whether to perform a kidney biopsy to make the diagnosis is individualized in each case; however, in the absence of proteinuria and/or hypertension and in light of the lack of proven therapies for IgA nephropathy, biopsies are less likely to be performed in children with microscopic hematuria versus those with recurrent gross hematuria. The biopsy specimen will demonstrate variable degrees of mesangial cell proliferation and IgA deposits on immunofluorescent staining. Hereditary nephritis also manifests clinically in childhood with either persistent microscopic hematuria or recurrent episodes of gross hematuria during upper respiratory infections. The key clinical step in distinguishing IgA nephropathy from hereditary nephritis is the absence of a family history of significant kidney disease in the former while there is a clear history of ESRD and hearing loss in first-order relatives of a child with potential hereditary nephritis. Hereditary nephritis spans a spectrum of entities all of which are the result of genetic mutations in type IV collagen genes that are constituents of the GBM (Kashtan, 2004). In benign familial nephritis or familial thin basement nephropathy the genetic mutation is in the α3 or α4 chains of type IV collagen, which are on chromosome 2 (Kashtan, 2004). In the heterozygous state the renal involvement is mild and the sole manifestation of disease is persistent microscopic hematuria. The risk of progressive loss of kidney function is generally low in these cases. In contrast, Alport syndrome, the most severe variant of hereditary nephritis, is caused by mutations in the α5 chain of type IV collagen on the X chromosome. This accounts for the more severe disease in males. The phenotype is more variable in females because of the Lyon hypothesis and random inactivation of an X chromosome in each cell. There are autosomal dominant and recessive forms of Alport syndrome linked to mutations in the α3 and α4 chains of type IV collagen. There are extrarenal manifestations of Alport syndrome (eye, platelets, smooth muscle) that can facilitate the diagnosis but, in general, the diagnosis is made based on characteristic ultrastructural changes in the GBM—splitting and lamellation on electron microscopic examination. Immunohistochemical staining for the various α chains of type IV collagen at the dermal-epidermal junction in a skin biopsy specimen can show alterations that are diagnostic of the various categories of hereditary nephritis (Kashtan, 2004). The use of genetic testing for Alport syndrome is becoming more accessible but is still not considered a routine procedure that can replace a kidney biopsy because identification of the unique mutation in most families is a costly and time-consuming procedure. The major renal manifestation of SLE is glomerular disease. The primary symptoms leading to the diagnosis of renal disease in children with SLE are proteinuria, microscopic hematuria, hypertension, and mild renal insufficiency (Cameron, 1994). SLE is an autoimmune disease with the potential to affect numerous organ systems. Children with SLE are more likely than adults to have renal involvement. The peak age at onset is 14 years, around the time of puberty, and females are more likely to be affected than males (M : F = 4.4 : 1). Half of children with lupus nephritis have nephrotic syndrome and/or renal insufficiency at diagnosis. SLE is divided into classes based on histopathology (the prevalence of each from several biopsy series in children is given in parentheses): class I, normal (6%); class II, mesangial proliferative (19%); class III, focal endocapillary proliferative (23%); class IV, diffuse endocapillary proliferative (43%); and class V, membranous (9%). Both C3 and C4 are commonly decreased during acute illness. Abnormalities common at presentation in children with lupus nephritis are fever (78%), arthralgia (75%), rash (68%), weight loss (40%), Coombs-positive anemia (48%), leukopenia (47%), thrombocytopenia (25%), coagulopathy (27%), and renal disease (82%). Hemolytic uremic syndrome is defined by the triad of microangiopathic hemolytic anemia, thrombocytopenia, and acute renal insufficiency. In children, hemolytic uremic syndrome may be the typical form, which is associated with diarrhea, or an atypical form (Pickering et al, 1994; Loirat and Taylor, 2004). This discussion will focus on the typical diarrhea-associated hemolytic uremic syndrome, which in North America is secondary to primary infection with enterotoxic Escherichia coli. The enteropathic E. coli infection is usually contracted through ingestion of food contaminated with bovine feces: ground beef, water supplies, or vegetables/fruits. Person-to-person spread is also possible. Specific serotypes of E. coli (O157:H7) produce a verotoxin that binds to enterocytes and gains access to the microcirculation. After endocytosis into vessels the toxin causes endothelial injury, particularly in the kidney, by inhibition of protein synthesis by the endothelial cell. Platelet-fibrin thrombi within glomeruli cause ischemia of glomerular capillaries associated with mesangiolysis. Severe forms are also characterized by cortical necrosis as larger arterioles may become involved. Key Points Glomerular Disease
Transition from the Fetus to the Infant
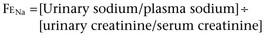
Principles of Fluids and Electrolytes
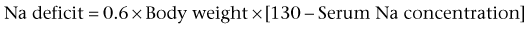

Hematuria
GROSS
MICROSCOPIC
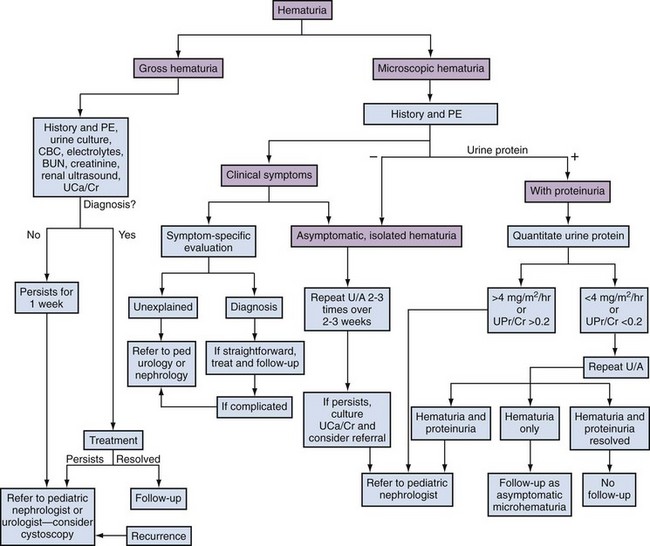
Epidemiology
Evaluation
Hypercalciuria
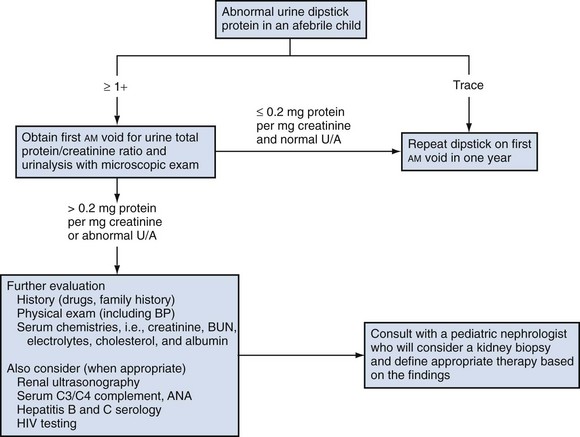
Proteinuria
Glomerular Disease
Nephrotic Syndrome
Nephrotic Syndrome in Infants
Minimal Change Nephrotic Syndrome
Focal Segmental Glomerulosclerosis
Postinfectious Glomerulonephritis
IgA Nephropathy
Hereditary Nephritis (Alport Syndrome)
Lupus Nephritis
Hemolytic Uremic Syndrome
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Prostate Cancer Tumor Markers
Prostate Cancer Tumor Markers
 Core Principles of Perioperative Management in Children
Core Principles of Perioperative Management in Children
 Neuropathic Dysfunction of the Lower Urinary Tract
Neuropathic Dysfunction of the Lower Urinary Tract
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Renal Functional Development and Diseases in Children
• Nephrotic syndrome is defined as edema, hyperlipidemia, hypoalbuminemia, and heavy proteinuria (>40 mg/m2/hr).
