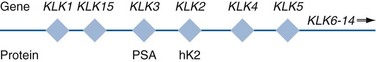
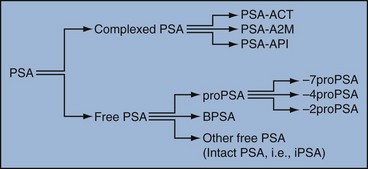
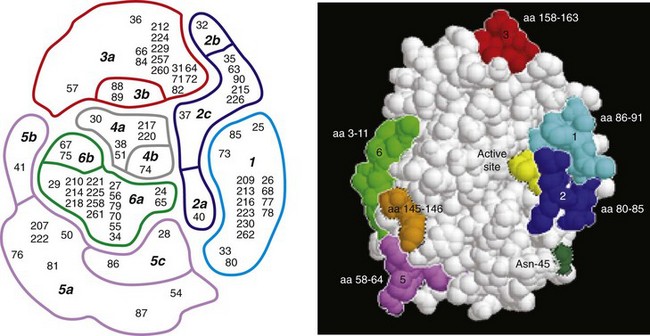
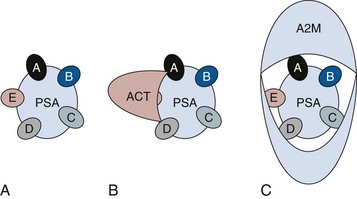
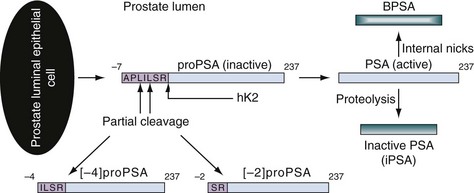
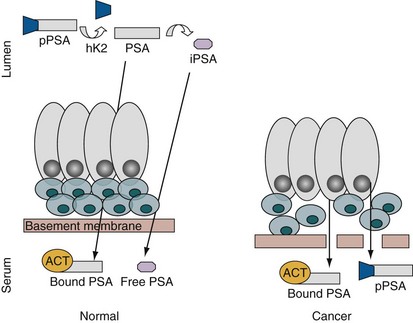
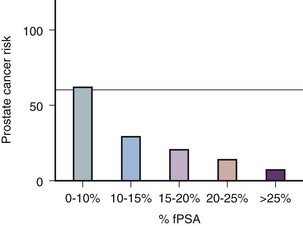
Robert H. Getzenberg, PhD, Alan W. Partin, MD, PhD Among urologic malignancies, prostate cancer has greatly benefited from the discovery and application of tumor markers. Since its discovery in 1979 until clinical application in the late 1980s through 1990s, the prostate-specific antigen (PSA) has evolved into an invaluable tool for detecting, staging, and monitoring prostate cancer in men (Sensabaugh, 1978; Wang et al, 1979, 1981; Papsidero et al, 1980; Kuriyama et al, 1981). The widespread use of PSA screening has generated greater awareness about prostate cancer. During the PSA era, cancers identified when confined to the prostate have improved cure rates using either radical prostatectomy or radiation therapy. Whereas the majority of prostate cancers in the 1980s and early 1990s commonly arose through finding an abnormal digital rectal examination (DRE) or elevated PSA, or both, today most prostate cancer arises as clinically nonpalpable (stage T1c) disease with PSA levels between 2.5 and 10 ng/mL. The evolving demographics and natural history of prostate cancer have resulted in a stage migration to nonpalpable, clinically localized (stage T1c) disease and a parallel reduction in mortality (Pound et al, 1997, 1999; Stephenson et al, 1997; Polascik et al, 1999). However, these PSA-detected T1c cancers are not homogeneous. Although PSA screening has improved survival, outcomes are not the same for all T1c-detected disease because some of these cancers may not pose a threat to survival (Gretzer et al, 2002). Methods for improved detection of clinically significant prostate cancer are needed. Despite routine application of PSA assays, limitations of specificity for this marker remain. Although PSA is widely accepted as a prostate cancer tumor marker, it is organ specific and not disease specific. Unfortunately, there is an overlap in the serum PSA levels among men with cancer and those with benign disease. Thus elevated serum PSA levels may reflect alterations within the prostate secondary to tissue architectural changes such as cancer, inflammation, or benign prostatic hyperplasia (BPH). Currently, serum PSA levels as low as 2.6 ng/mL are used as a threshold to perform transrectal ultrasound-guided biopsy. Although up to 30% of men presenting with an elevated PSA may be diagnosed following this invasive procedure, as many as 75% to 80% are not found to have cancer. In some instances the biopsy needle may fail to sample representative areas, thus failing to detect present cancer. To this end, application of PSA derivatives such as PSA density, PSA velocity, age-adjusted values, and, more recently, molecular derivatives have attempted to improve the performance of PSA (Christensson et al, 1990; Benson et al, 1992a, 1992b; Carter et al, 1992a, 1992b; Oesterling et al, 1993a, 1993b, 1993c; Riehmann et al, 1993; Seaman et al, 1993; Bazinet et al, 1994; Rommel et al, 1994; McCormack et al, 1995; Morgan et al, 1996; Lilja, 1997). Changes in gene expression may occur as a result of alterations in DNA. Alterations known as epigenetic modifications include changes in DNA methylation and histone acetylation status. Segments within the gene promoter that are composed of GC-rich regions are termed CpG islands. Alterations in the methylation status of these regions may affect gene expression and have been shown to play a role in carcinogenesis (Jones and Baylin, 2002). Furthermore, cumulative effects of environmental exposures, such as diet and stress throughout life, may affect DNA methylation status and thus contribute to the risk of cancer development (Li et al, 2004). Assays for detection of CpG island hypermethylation include Southern blotting, restriction endonuclease PCR (RE-PCR), bisulfite genomic sequencing (BCR), and methylation-specific (MSP) PCR. Although each assay is sensitive, technical limitations exist. Currently, MSP PCR is most commonly employed for the detection of methylated CpG islands (Jones, 2002). The products of two hypermethylated genes that have been evaluated in prostate cancer development are glutathione-S-transferase P1 (GSTP1) and RAS association domain family protein isoform A (RASSF1A). GSTP1 belongs to a family of detoxifying enzymes that are involved in metabolic reduction of electrophilic carcinogens. These enzymes are thought to be involved in the development of prostate cancer. Elevated levels of GSTP1 CpG hypermethylation have been detected in tissues from precancerous lesions (atypia and prostatic intraepithelial neoplasia [PIN]) and within ejaculates, urine, and plasma from men with prostate cancer (Nakayama et al, 2003). To date, many studies have evaluated these hypermethylated CpG islands as a prostate cancer tumor marker (Cairns et al, 2001; Goessl et al, 2001a, 2001b; Gonzalgo et al, 2003; Bastian et al, 2004). Cairns and coworkers (2001) have demonstrated the presence of elevated GSTP1 hypermethylation in up to 79% of prostate cancer specimens. Using urine specimens obtained after prostate massage, Goessl and colleagues (2001a, 2001b) found elevated levels of this marker in 68% of men with organ-confined disease, 78% of men with locally advanced or metastatic disease, 29% of men with PIN, and 2% of men with BPH. Gonzalgo and coworkers (2003) demonstrated elevated GSTP1 hypermethylation in up to 50% of urine sediments taken from men immediately after transrectal prostate biopsy. Hypermethylation was also detected in 33% of men with negative biopsies and in up to 67% of men found to have PIN or atypia, suggesting that these patients may harbor occult prostate cancer and require more rigid follow-up with a low threshold for repeat biopsy. The use of MSP to measure GSTP1 hypermethylation in body fluids represents an emerging tool that is feasible and has demonstrated reproducible results, and it is likely to become a valued screening modality for prostate cancer detection. In addition to GSTP1, hypermethylation of RASSF1A has been noted to occur in up to 70% of prostate cancers (Kuzmin et al, 2002; Liu et al, 2002). Early findings have noted an association of this marker with more aggressive tumors, and it may aid in distinguishing between these cancers and more indolent cancers. Although early, this work is promising, and the subject remains under investigation. Many men continue to undergo biopsy procedures even though only 10% to 36% of men having an initial negative biopsy are found to have cancer on second biopsy (Djavan et al, 2000; O’Dowd et al, 2000; Lopez-Corona et al, 2003; Singh et al, 2004; Eggener et al, 2005), and cancer detection decreases with each subsequent negative biopsy (Keetch et al, 1994). Improved methods are needed to identify men who can forgo a repeat biopsy, particularly if the trigger for initial biopsy or the initial biopsy histology indicates high-risk features. Therefore emphasis on achieving a high negative predictive value (NPV) and low false-negative rate has been championed following many biomarker evaluations. In initial studies of prostate cancer, DNA methylation markers have exhibited high sensitivity and specificity (Maruyama et al, 2002; Yegnasubramanian et al, 2004), and improved upon the sensitivity of histology alone by 15% (Harden et al, 2003a). We therefore conducted this study to evaluate whether DNA methylation markers could improve upon the NPV of histology alone in men without cancer on initial biopsy but with a high index of suspicion for a missed cancer. The first tissue methylation-biomarker to be discovered was glutathione-S-transferase π (GSTP1). Glutathione S-transferases (GSTs) are a family of detoxification enzymes that conjugate reactive substrates with reduced glutathione. The enzymes are dimers classified into four groups: α, µ, π, and θ (Hayes et al, 1995). Li and coworkers (2004) noted that the glutathione-S-transferase π (GSTP1) gene was unmethylated in all normal human tissues and BPH but was hypermethylated in all 20 prostate cancer (CaP) specimens analyzed. Eighty-eight of 91 CaP specimens did not stain for GSTP1 protein. Harden and coworkers (2003a, 2003b) used PCR to detect hypermethylated GSTP1 in prostate biopsy specimens. Twenty-nine (of 45 patients) were able to be evaluated using this method. GSTP1 methylation was detected in 11 of 15 (73% sensitivity) CaP cases and in none of 14 (100% specificity) benign controls. Quantitation of GSTP1 hypermethylation accurately detected CaP even in small, limited tissue samples. This would indicate that methylated GSTP1 represents a marker useful for CaP screening. Bastian and coworkers (2005) used a restriction endonuclease, QMSP (RE-MSP), to detect abnormalities in the CpG islands found in serum GSTP1 DNA. No men with a negative biopsy had GSTP1 DNA detected in their sera, compared with 12% of men with clinically localized CaP and 28% with metastatic disease. With the landmark discovery of the presence of gene fusions in prostate cancer by Chinnaiyan and colleagues (Tomlins et al, 2005), considerable work has been performed to determine if assays can be developed that can accurately detect prostate cancer or provide prognostic information. The TMPRSS2 and ETS transcription factor fusions have revealed novel biology about solid tumors with great potential implication (Tomlins et al, 2005). Because this discovery has such great potential, the concept has been to determine if we can detect these fusions in the urine of men with prostate cancer (Barry et al, 2007). A feasibility study that examined urine samples post-DRE, revealed that indeed the TMPRSS2-ERG fusion can indeed be found within the urine and has the potential to be an interesting marker (Laxman et al, 2008) with potential to aid in the detection of prostate cancer in men about to undergo a biopsy for the disease. In a larger follow-up study, investigators were able to determine that gene fusions are detectable in approximately 37% of the urine samples obtained from men with prostate cancer (Hessels et al, 2007). Using tissue samples, the presence of these gene fusions was observed in about half of the prostate cancer cases and was, in fact, observed in an even lower percentage of the more aggressive tumors (Perner et al, 2007; Mosquera et al, 2008). Several assays are being developed to detect these fusions in tissue and urine samples (Lu et al, 2008). Further delineation of the various splice variants that have been produced has the potential to add to the diagnostic utility of these fusions, indicating that the detection of a single fusion may not be sufficient (Hu et al, 2008). The clinical utility of the detection of a single gene fusion alone has been questioned recently with findings that the TMPRSS2-ERG fusion does not correlate with outcome in men undergoing prostatectomies (Gopalan et al, 2009). Moving toward the concept that single biomarkers will probably not answer the important clinical questions in prostate cancer alone, many investigators have begun to multiplex markers, resulting in additive value. Combining both the prostate cancer–3 (PCA-3) test, that will be described later in the chapter, together with the detection of these gene fusions in urine resulted in increasing sensitivity to 73%, although it was not possible to determine the specificity of the combined tests in these clinical populations. Overall, the current data related to the use of urine-based gene fusions seems to indicate that, at best, they may add slightly to PSA or even PCA-3 detection of prostate cancer and does not appear to have the ability to distinguish aggressive from nonaggressive disease. As the genetic alterations associated with disease states are mapped, novel information is being revealed related to the potential of these differences to serve as biomarkers for prostate cancer. These findings have been specifically associated with more advanced prostate cancer. Using arrays that can specifically analyze single nucleotide polymorphisms (SNPs), investigators have begun to perform large-scale genomic comparisons of genetic alterations associated with prostate cancer. Much of the initial work has been done on individuals who have family histories of prostate cancer. In work performed through a collaborative effort that examined large numbers of individuals who are part of these hereditary study cohorts, investigators have revealed SNP patterns that impart a higher risk for developing prostate cancer. Because these are genomic differences, a simple blood draw or cheek swab can be used to make such a determination (Sun et al, 2008). Although the data are quite dramatic, the question has been raised about how these data should be handled clinically. For example, if an individual is found to have a fivefold increase in their risk for prostate cancer, what should be done? Should they be screened more carefully or more frequently? Should they simply undergo a prostate biopsy or treatment? Certainly this seems like an application where some type of preventive strategy might work. Unfortunately, we have little, if anything available that has been used to effectively prevent the disease. In an attempt to discern genomic polymorphisms that may be predictive of castrate-resistant prostate cancer, Luo and colleagues (Hu et al, 2009) have examined the androgen receptor to determine if there may be gene variants that are predictive of the ability of the tumor to continue to grow in the absence of androgens. The hypothesis is that there are distinct forms of the androgen receptor that may be active in the absence of ligands. This may permit the cancer cells to continue to grow even in a castrate environment. They have recently revealed the presence of several unique forms of the androgen receptor. One form, variant 7, is shown to be increased in men with advanced prostate cancers (Hu et al, 2009). Using an antibody being constructed, the researchers are proposing to develop a blood test that can be used to identify individuals with castrate- resistant prostate cancer. The glycoprotein prostate-specific membrane antigen (PSMA) is a folate hydrolase and is found embedded within the cell membrane of all prostatic epithelial cells. The hydrophobic amino acids found on amino acid residues 20 to 43 suggest that this protein is a type II integral membrane protein with a small intracellular domain and a large extracellular domain (Israeli et al, 1997; Fair et al, 1997). The gene for PSMA has been cloned, fully sequenced, and localized to the short arm of chromosome 11 (11p11-p12). PSMA has been identified in the central nervous system, intestine, and prostate. In the brain, PSMA functions to metabolize the neurotransmitter N-acetyl-aspartyl-glutamate. In the intestine, PSMA has been localized to the proximal small bowel, where it works as a carboxypeptidase. Although isolated in these other tissues, PSMA is predominantly expressed in the prostate gland. Of interest for diagnosis (Douglas et al, 1997), prognosis (Perner et al, 2007), and imaging (Wynant et al, 1991; reviewed by Abdel-Aziz et al, 2000) is the discovery of elevated expression of this protein in tissue from prostate cancer compared with normal prostate tissue (Silver et al, 1997; Chang et al, 1999a; Elgamal et al, 2000). More work has focused on use of PSMA in treatment strategies than for diagnostic tests (Elsässer-Beile et al, 2009). Multiple monoclonal antibodies have been designed to identify both the intracellular and extracellular domains of the PSMA protein; however, few have shown “diagnostic promise” immunohistochemically or as serum/urine assays (Horoszewicz et al, 1987; Douglas et al, 1997). However, PSMA mRNA expression among prostate cancers is highest in the hormone-deprived state (Henttu et al, 1992; Israeli et al, 1994). During cancer progression, differentially expressed variants of PSMA have been identified. Of three alternatively spliced variants of PSMA, one, known as PSM′, is differentially expressed in normal tissue, BPH, and prostate cancer. Su and colleagues (1995) demonstrated that the PSMA/PSM′ ratio is upregulated three- to sixfold in prostate cancer compared with BPH (0.76 to 1.6) and normal (0.075 to 0.45) tissue. Further reports to date have demonstrated the production of antibodies to PSMA. Horoszewicz and coworkers (1987) have found increased PSMA levels in 47% of prostate cancer patients (20 of 43) versus only 5% of noncancer patients (3 of 66) and negative results in 30 normal blood donors. Using the antibody produced by Horoszewicz and coworkers, Xiao and colleagues (2001) reported use of an immuno-SELDI (surface-enhanced laser desorption ionization) assay for PSMA. Applying the antibody to PSMA on a ProteinChip array, these authors were able to differentiate cases of prostate cancer from BPH. Perner and colleagues (2007) demonstrated marked differences, immunohistochemically, in the levels of PSMA in tissues from various prostate tumors that progressed after surgery (HR = 1.5, 95% CI = 1.1 to 2.8, P = .17) and suggested that PSMA might act independently as a prognostic biomarker of disease progression. The development of an accurate enzyme-linked immunosorbent assay (ELISA) for PSMA in serum/urine has potential significance for diagnostic and prognostic evaluation for prostate cancer. The clinical usefulness of PSMA for diagnosis, monitoring, and imaging continues to be promising and remains under investigation in many laboratories. Another source of blood-based prostate cancer biomarkers has been identified among the protein products of the human kallikrein gene family (Fig. 98–1). Originally, only three genes of this family of genes were identified: the pancreatic/renal kallikrein (hKLK1), human kallikrein 2 (hKLK2), and PSA (hKLK3) genes (McCormack et al, 1995; Rittenhouse et al, 1998; Diamandis et al, 2000; Lilja, 1997). Since the identification of 12 other kallikrein genes, this family of proteases now consists of 15 members and is described with a distinct nomenclature (Diamandis et al, 2000; Yousef and Diamandis, 2001). Located on the long arm of chromosome 19 within the region spanning q13.2-q13.4, these genes share similar structural similarities. The serine proteases encoded by the three originally described genes have been identified as hK1 or hPRK (KLK1 gene), hK2 or hGK-1 (KLK2 gene), and hK3 or PSA (KLK3 gene). These serine proteases have similar amino acid sequences, with hK1 expressing 60% and hK2 expressing 78% homology with PSA (Schedlich et al, 1987; Clements, 1989). Both hK2 and hK3 are released in zymogen form from the prostatic epithelium and are found in seminal fluid as well as serum. Because they share structural homology, they can both form complexes with endogenous protease inhibitors such as α2-macroglobulin and α1-antichymotrypsin (ACT) (Young et al, 1995; Lilja, 1997; Rittenhouse et al, 1998). hK2 has been shown to regulate PSA activity by cleaving a leader amino acid sequence from PSA (proPSA), thus activating PSA (Kumar et al, 1997; Lovgren et al, 1997; Takayama et al, 1997). Although PSA is less expressed in prostate cancer tissue, hK2 levels have been shown to be elevated in poorly differentiated prostate cancer (Darson et al, 1997; Tremblay et al, 1997). Similar to PSA, hK2 has been shown to aid in the detection of prostate cancer (Kwiatkowski et al, 1998; Partin et al, 1999; Nam et al, 2000). It is likely that several other members of the kallikrein family of serine proteases will become useful tumor markers. Both hK6 and hK10 have been described as potential serum biomarkers for nonprostatic diseases, especially ovarian cancer (Luo et al, 2001; Yousef and Diamandis, 2001). The most notable marker in the kallikrein family is hK3, also known as PSA. It was first identified and purified in the late 1970s, but widespread use in clinical urology did not occur for another decade (Ablin et al, 1970; Sensabaugh, 1978; Wang, 1979; Papsidero, 1980; Kuriyama, 1980, 1981; Wang, 1981; Seamonds, 1986; Chan et al, 1987; Stamey et al, 1987; Oesterling et al, 1988). PSA is a 33-kD glycoprotein that acts as a serine protease. The ectopic expression of PSA has been reported in smaller concentrations in the tissue of malignant breast tumors (Yu et al, 1994a, 1994b, 1994c), normal breast tissue (Monne et al, 1994; Yu et al, 1995), breast milk (Yu and Diamandis, 1995), female serum (Yu et al, 1995), and adrenal and renal carcinomas (Levesque et al, 1995a); however, for practical and clinical purposes, PSA is organ specific, primarily produced by the prostatic luminal epithelial cells (Yu et al, 1995; Levesque et al, 1995b; Diamandis et al, 2000). Although it is organ specific, PSA is not cancer specific, as demonstrated by the substantial overlap in values between men with benign versus malignant prostate disease (Oesterling et al, 1988; Partin et al, 1990). The function of this androgen-regulated protease is to liquefy semen through its action on the gel-forming proteins semenogelin and fibronectin within the semen following ejaculation (Lilja and Laurell, 1984; Lilja, 1985; Lilja et al, 1987; McGee and Herr, 1988; Christensson et al, 1990). PSA is normally found in low concentration in sera (ng/mL). Within sera, PSA circulates in both bound and unbound forms (Fig. 98–2). Most PSA in sera is bound or complexed to the antiproteases ACT and macroglobulin (MG) (Christensson et al, 1990; Lilja et al, 1991; Stenman et al, 1991). Binding of free PSA to ACT inactivates the protease, but the complex PSA-ACT remains immunodetectable by current assays (Partin et al, 2003). Binding of PSA to MG still allows some proteolytic activity but renders the PSA-MG complex undetectable by most current assays (Christensson et al, 1990). Free PSA without proteolytic activity is probably rendered inactive within the prostatic epithelial cell before release into the sera. This free inactive PSA does not form complexes with antiproteases, circulates unbound in sera, and is immunodetectable by current assays (Lilja et al, 1991). The primary release of PSA into the seminal fluid results in 106-fold higher seminal concentrations than levels measured within serum (Sensabaugh, 1978; Wang et al, 1981; Lilja and Abrahamsson, 1988; McCormack et al, 1995). The concentrations found in seminal plasma range from 0.5 to 5.0 mg/mL, whereas the normal serum concentration in men aged 50 to 80 years without prostate disease ranges between 1.0 and 4.0 ng/mL (Catalona et al, 1991). Prostate cancer cells do not necessarily make more PSA than do normal prostate cells, and elevated serum levels are probably a result of cancer progression and destabilization of the prostate histologic architecture (Stamey et al, 1987). Studies have demonstrated that prostate cancer cells do not make more PSA but rather make less PSA than normal prostatic tissue (Meng et al, 2002). Evaluation of tissue from prostate cancer specimens has demonstrated up to 1.5-fold lower messenger RNA (mRNA) expression levels compared with normal prostate tissue (Meng et al, 2002). PSA expression is strongly influenced by androgens (Young et al, 1991; Henttu et al, 1992). Immunohistochemical detection of PSA within the prostate is characterized by bimodal peaks between 0 and 6 months and after 10 years, correlating directly with testosterone levels (Goldfarb, 1986). Serum PSA becomes detectable at puberty with increases in luteinizing hormone and testosterone (Vieira et al, 1994). In the absence of prostate cancer, serum PSA levels vary with age, race, and prostate volume. In men without BPH, the rate of change in PSA is 0.04 ng/mL per year (Carter et al, 1992b; Oesterling, 1993), compared with 0.07 to 0.27 ng/mL per year in men with BPH who are between the ages of 60 and 85 years (Carter et al, 1992b). Cross-sectional data suggest that PSA increases 4% per milliliter of prostate volume and that 30% and 5% of the variance in PSA can be accounted for by prostate volume and age, respectively (Oesterling et al, 1993a). Blacks without prostate cancer have higher PSA values than whites (Morgan et al, 1996; Fowler et al, 1999). Fowler and colleagues (1999) have demonstrated that, on a volume/volume basis, the benign prostatic tissue of black men contributes more PSA to sera than does the benign prostatic tissue of white men, a difference that increases with age. Elevated serum PSA levels are probably a product of disruption of cellular architecture within the prostate gland (Stamey et al, 1987). The loss of the barrier afforded by the basal layer and basement membranes within the normal gland is a likely site for the egress of PSA into the circulation. This can occur in the setting of prostate disease (BPH, prostatitis, prostate cancer) and with prostate manipulation (prostate massage, prostate biopsy) (Stamey et al, 1987). Prostatic inflammation (acute and chronic) and urinary retention can cause PSA elevations to variable degrees (Armitage et al, 1988; Dalton, 1989; Nadler et al, 1995). PSA elevations may not be related to the histologic finding of inflammation in men without clinical prostatitis (Morote et al, 2000). Prostatic trauma, such as occurs after prostatic biopsy, can result in a leak of PSA into the circulation that may require more than 4 weeks for return to baseline values (Yuan et al, 1992). DRE as performed in an outpatient setting can lead to increases in serum PSA (Thompson and Zeidman, 1992). However, the change in PSA after DRE does not appear to be clinically significant, because the change is within the error of the assay and rarely causes false-positive tests (Chybowski et al, 1992; Crawford et al, 1992; Thompson and Zeidman, 1992). Studies of the effect of ejaculation on serum PSA have shown both no significant change in PSA (Kirkali et al, 1995) and a significant decrease in serum PSA (Simak et al, 1993; Heidenreich et al, 1997) in men 30 to 40 years old or younger. However, in the age group in which PSA testing is primarily used for early detection of prostate cancer (50 years and older), ejaculation can lead to an increase in PSA that can result in a false-positive elevation (Tchetgen et al, 1996; Herschman et al, 1997). After 48 hours, this fractional increase in PSA would be expected to return to baseline levels in most men (Tchetgen et al, 1996). A history of sexual activity and a repeated PSA test after 48 hours of sexual abstinence may be helpful in the interpretation of serum PSA levels that are minimally elevated. The presence of prostate disease (prostate cancer, BPH, and prostatitis) is the most important factor affecting serum levels of PSA (Wang et al, 1981; Ercole et al, 1987; Robles et al, 1988). PSA elevations may indicate the presence of prostate disease, but not all men with prostate disease have elevated PSA levels. Furthermore, PSA elevations are not always specific for prostate cancer. Prostate-directed treatment (for both BPH and cancer) can lower serum PSA by decreasing the volume of prostatic epithelium available for PSA production and by decreasing the amount of PSA produced per cell. Manipulation of the hormonal environment for treatment of cancer and BPH with orchiectomy, luteinizing hormone–releasing hormone analogs, and 5α-reductase inhibition; radiotherapy; and surgery for BPH or cancer can all lead to reductions in serum PSA (Henttu and Vihko, 1992). Finasteride (5 mg) and other 5α-reductase inhibitors (dutasteride) for treatment of BPH have been shown to lower PSA levels by an average of 50% after 6 months of treatment (Guess, 1993). Thus one can multiply the PSA level by two to obtain the “expected” PSA level of a patient who has been taking a 5α-reductase inhibitor—for 6 months or more. Men who are to be treated with a 5α-reductase inhibitor should have a baseline PSA measurement before initiation of treatment and should be observed with serial PSA measurements, because this drug may decrease PSA serum levels by up to 50%. If PSA does not decrease by 50% or if there is a rise in PSA when the patient is taking a 5α-reductase inhibitor, the patient should be suspected of having occult prostate cancer and undergo further testing. Finasteride (1 mg), also marketed for the treatment of male baldness, may also lower PSA levels to a lesser extent than the 5-mg dosage. Other over-the-counter medications and neutraceutical use should always be documented. Although saw palmetto has not been shown to affect PSA levels, these unregulated supplements may contain compounds that can alter PSA levels (e.g., PC-SPES, now off the market). Interpretation of PSA values should always take into account the presence of prostate disease, previous diagnostic procedures, and prostate-directed treatments. One of the most exciting and clinically useful discoveries since the introduction of PSA testing has been the demonstration of different molecular forms of PSA in serum (Christensson and Lilja, 1994; Christensson et al, 1990, 1993; Lilja et al, 1991; Stenman et al, 1991; McCormack et al, 1995; Lilja et al, 1997; Polascik et al, 1999). Measurable PSA found circulating in the blood exists either complexed (bound, cPSA) to proteins or as a free (unbound) form (fPSA) (see Fig. 98–2). Three proteins that are known to bind to PSA in the blood are ACT, α2-macroglobulin (A2M), and α1-protease inhibitor (API) (Christensson et al, 1990, 1993, 1994; Lilja et al, 1991; McCormack et al, 1995; Tewari et al, 1996; Lilja, 1997). Figure 98–3 illustrates the molecular model of PSA and its epitope sites. The majority of PSA that enters the serum is bound (70%) to these proteins. When bound to A2M, all PSA epitope sites become masked, making this complex difficult to measure (Fig. 98–4). Of the complexed PSA derivatives found in serum, PSA bound to ACT (PSA-ACT) is immunoreactive and found in the greatest concentration. Although representing a lower proportion of total PSA in the blood, fPSA is also immunoreactive and therefore measurable. Development of new monoclonal antibodies specific for fPSA and cPSA has allowed more accurate measurement of the different molecular forms of PSA and their ratios in serum (Table 98–1) (Lilja et al, 1991; Stenman et al, 1991). Table 98–1 Molecular Derivatives of Prostate-Specific Antigen ACT, α1-antichymotrypsin; API, α1-protease inhibitor; A2M, α2-macroglobulin; PSA, prostate-specific antigen. Although the majority of serum PSA is found complexed to the proteases (most to ACT), 5% to 35% of PSA exists as fPSA (McCormack et al, 1995; Tewari et al, 1996; Woodrum et al, 1998). Although prostate cancer cells do not produce more PSA than benign prostate epithelium, the PSA produced from malignant cells appears to escape proteolytic processing. Thus men with prostate cancer have a greater fraction of serum PSA complexed to ACT and a lower percentage of total PSA that is free compared with men without prostate cancer (Christensson et al, 1993; Leinonen et al, 1993; Lilja, 1993; Stenman et al, 1994; Catalona et al, 1997). Using this important observation led to the measurement of the percentage of the ratio of free to total PSA (%fPSA) and has since provided an additional degree of specificity for prostate cancer detection (Catalona et al, 1998; Partin et al, 1998). The difference in the ratio of free to total PSA (percentage of free PSA or %fPSA) is greatest when comparing men without prostate cancer who have prostate enlargement (BPH) with those with prostate cancer and no prostate enlargement. This difference may be due to differential expression of PSA isoforms by transition zone (zone of origin of BPH) tissue compared with peripheral zone (zone of most prostate cancer) tissue (Chen et al, 1997; Mikolajczyk et al, 2000). The role for %fPSA is more applicable to PSA levels less than 10 ng/mL, because the positive predictive rate of total PSA above 10 to 20 ng/mL has been demonstrated to be as high as 80%. Currently, %fPSA is approved by the Food and Drug Administration (FDA) for use to aid PSA testing in men with benign DRE and minimal PSA elevations, within the diagnostic gray zone of 4 to 10 ng/mL. If PSA levels are less than 4.0 ng/mL, more recent data have demonstrated the utility of %fPSA when deciding whether a patient requires an initial or repeat biopsy. Christensson and coworkers (1993) measured free and total PSA fractions in men with and without prostate cancer and found that a free/total PSA cutoff of 0.18 (18% free/total PSA) significantly improved the ability to distinguish between subjects with and without cancer compared with use of total PSA alone. Other %fPSA cutoff values have also been identified to improve risk stratification. Catalona and colleagues (1995) have found that %fPSA provided independent predictive information regarding the presence or absence of cancer above that provided by other clinical indices, including age, total PSA, DRE results, and prostate size. As many as 20% to 65% of unnecessary biopsies may be avoided when using %fPSA cutoff values ranging between 14% and 28%, while maintaining sensitivity rates from 70% to 95% within the tPSA range of 4 to 10 ng/mL (Catalona et al, 1998; Partin et al, 1998; Veltri and Miller, 1999; Vessella et al, 2000; Stephan et al, 2002). In a prospective, multi-institutional study of men 50 to 75 years old with PSA levels between 4 and 10 ng/mL and palpably benign prostate glands, a %fPSA cutoff of 25% (biopsy less than 25%) detected 95% of cancers while avoiding 20% of unnecessary biopsies (Catalona et al, 1998). These data confirm that the application of %fPSA can help counsel men with PSA elevation between 4 and 10 ng/mL regarding their risk of cancer and the need for further evaluation to rule out the disease (Fig. 98–5). (From Catalona WJ, Partin AW, Slawin KM. Percentage of free PSA in black versus white men for detection and staging of prostate cancer: a prospective multicenter clinical trial. Urology 2000;55:372–6.) A number of other studies have demonstrated the usefulness of %fPSA in prostate cancer detection (for reviews see Polascik et al, 1999; Karazanashvili et al, 2003). Increased use of PSA as a screening tool has led to increased interest in the evaluation of PSA levels below 4.0 ng/mL. Examination of cancers detected within the PSA range 2.5 to 4.0 ng/mL has shown numbers of cancers similar to those detected in the 4 to 10 ng/mL range and also that these cancers are clinically significant and may exhibit aggressive pathologic potential (Colberg et al, 1993; Smith et al, 1998; Schroder et al, 2000; Recker et al, 2001). Djavan and colleagues (1999) demonstrated that for men with PSA levels less than 4 ng/mL, a %fPSA cutoff value of 27% could detect up to 90% of cancers while preventing 18% of unnecessary biopsies. Furthermore, comparison against PSA derivatives, such as prostate-specific antigen density (PSAD), PSA velocity, and transition zone density favored %fPSA. Haese and coworkers (1997) demonstrated that %fPSA in the total PSA (tPSA) range of 2 to 4.0 ng/mL does not substantially increase the number of biopsies needed to detect clinically significant prostate cancer compared with that in the 4 to 10 ng/mL range. In this study, a %fPSA cutoff value of 18% to 20% detected almost half of cancers while sparing 73% of men from undergoing biopsy, with a biopsy-to-cancer ratio of 3 : 1 to 4 : 1. One study has demonstrated that a %fPSA cutoff of 25% would lead to detection of 95% of prostate cancers in both white and black men (Catalona et al, 2000), suggesting that race may not be important when using %fPSA in cancer detection. Free PSA and total PSA both decrease in men receiving finasteride. As both decline, the percentage of fPSA is not altered significantly by this medication (Keetch et al, 1997; Pannek et al, 1998). In addition to contributing to detection, %fPSA has been shown to provide prognostic information. Serial measurement of %fPSA within archival serum demonstrated early marked and sustained differences in aggressive and nonaggressive prostate cancers (Carter et al, 1997). This group identified a statistically significant difference in %fPSA in men with and without metastatic disease up to 10 years prior to the development of prostate cancer. For the same cohort of men, total PSA levels failed to differentiate aggressive from nonaggressive prostate cancer 10 years prior to diagnosis. This study suggests that the longitudinal measurement of %fPSA changes may aid in detection and contribute information regarding disease behavior, thus providing useful information for therapeutic decision making. Despite these encouraging results, several important factors should be kept in mind with regard to interpretation of %fPSA in clinical practice for cancer detection (Meyer et al, 1997). Such factors include prostatic manipulation, specimen handling, and assay variation (Partin et al, 1996a, 1996b; Stephan et al, 1997; Roth et al, 1998; Woodrum et al, 1998). Prostatic manipulation and urethral instrumentation have been shown to affect the interpretation of the ratio of fPSA to total PSA. Although there may be marginal changes in total PSA, fPSA levels may fluctuate, affecting the %fPSA calculation. Furthermore, because fPSA is cleared more rapidly from serum than complexed PSA forms (i.e., total PSA), the resulting calculated %fPSA is directly affected. Thus it is recommended that PSA determinations be avoided for several weeks following prostatic manipulation (surgery, biopsy, cystoscopy) (Partin et al, 1996b; Lein et al, 1997; Bjork et al, 1998). Prostate volume has also been shown to influence the serum ratio of free to total PSA (Catalona et al, 1995; Partin et al, 1996; Haese et al, 1997; Stephan et al, 1997; Woodrum et al, 1998). The cutoff for %fPSA that optimizes sensitivity and specificity for cancer detection depends on prostate size because overlap in %fPSA is greatest among men without cancer who have enlarged prostates and men with cancer in the setting of prostate enlargement (Catalona et al, 1995). This group demonstrated that using a %fPSA cut point of 23% in men with PSA levels between 4 and 10 ng/mL and a prostate volume of less than 40 cm3 could yield 90% sensitivity. However, for men with larger prostates, the %fPSA cut point dropped to 14% in order to maintain this degree of sensitivity. The recommended and FDA-approved use for %fPSA remains for men with PSA levels within the diagnostic gray zone of 4 to 10 ng/mL. However, many clinicians use this ratio to aid in the decision making process regarding repeat biopsy (Stephan et al, 1997; Hayek et al, 1999; Djavan et al, 2000). For an initial biopsy, %fPSA ranges of 18% to 25% are commonly suggested; however, a widely accepted cutoff value has yet to be determined. For a repeat biopsy, Stephan and colleagues (1997) reported a 5% cancer miss rate when using a %fPSA value of 21% to trigger rebiopsy. A study by Hayek and coworkers (1999) illustrated a 16% positive rebiopsy rate regardless of %fPSA and concluded that this derivative was unable to provide further risk stratification for men in their population. Djavan and colleagues concluded that %fPSA is an accurate predictor of prostate cancer at rebiopsy. This group identified a %fPSA of 30% as the most accurate predictor of cancer in repeat biopsy specimens. However, until more series mature, the aggressiveness of this application in screening and follow-up biopsies should be tailored on a case-by-case basis (Luderer et al, 1995; Catalona, 1996; Partin et al, 1996b; Stephan et al, 1997; Catalona et al, 1998; Hayek et al, 1999; Djavan et al, 2000). Classification of free PSA isoforms can be done by assessing the presence or absence of internal cleavages, in which case the respective “forms” are termed “nicked” or “intact.” PSA originates with a 17–amino acid chain that is cleaved to yield a precursor inactive form of PSA termed proPSA (pPSA) (Zhang et al, 1995; Kumar et al, 1997, 2000; Mikolajczyk et al, 1997, 2001, 2002; Peter et al, 2001). As depicted in Figure 98–6, the precursor form of PSA contains a 7–amino acid proleader peptide, in addition to the 237 constituent amino acids of mature PSA, and is termed [–7]pPSA. Once released, the proleader amino acid chain is cleaved at the amino terminus by hK2, converting pPSA to its active 33-kD PSA form. In addition to hK2, pPSA may be activated to PSA by other prostate kallikreins, including hK4 (Takayama et al, 1997). Incomplete removal of the 7–amino acid leader chain has led to the identification of various other truncated or clipped forms of pPSA. These include pPSAs with 2, 4, and 5 leader amino acids ([–2]pPSA, [–4]pPSA, and [–5]pPSA) (Zhang et al, 1995; Kumar et al, 1997, 2000; Mikolajczyk et al, 1997, 2001, 2002; Peter et al, 2001). With cellular disruption, these inactive forms circulate as free PSA and may constitute the majority of the circulating free PSA in patients with prostate cancer (Mikolajczyk et al, 1997) (Fig. 98–7). Reports by Mikolajczyk and colleagues (2000, 2001)
Tissue Biomarkers
Glutathione-S-Transferase Pi (GSTP1)
Gene Fusions
Gene and Androgen Receptor Polymorphisms
Blood and Urine Biomarkers
Prostate-Specific Membrane Antigen
Human Kallikrein Gene Family
Prostate-Specific Antigen (PSA or hK3)
Molecular Derivatives of Prostate-Specific Antigen
PSA TYPE
% IN SERUM
Complexed PSA
60-95
PSA-ACT
60-90
PSA-API
1-5
PSA-A2M
10-20
Free PSA
5-40
Free PSA and the Percent Free PSA
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Core Principles of Perioperative Management in Children
Core Principles of Perioperative Management in Children
 Neuropathic Dysfunction of the Lower Urinary Tract
Neuropathic Dysfunction of the Lower Urinary Tract
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree