CHAPTER 4
Prostaglandins and other mucosal protecting agents
Introduction of drug class
The history of prostaglandins began in the 1930s with the observation by two New York gynecologists, Kurzrok and Lieb, that human semen caused contractions and relaxation of human myometrium. These observations were soon confirmed by Goldblatt in England and by von Euler in Sweden. Indeed, these compounds were so named because they were believed (incorrectly) to be derived from prostatic secretions. A crucial discovery, in terms of understanding the role of prostaglandins in the stomach, was the finding by Vane in 1971 that aspirin, indomethacin, and other nonsteroidal anti-inflammatory drugs (NSAIDs) inhibited the synthesis of prostaglandins. Vane suggested that the inhibition of prostaglandin biosynthesis by NSAIDs may underlie the ability of this class of drugs to induce ulceration in the gastrointestinal tract. Vane also proposed that this mechanism accounted for the anti-inflammatory properties of these compounds, since prostaglandins were known at the time to contribute to edema formation and to the pain associated with inflammation.
In the decade that followed Vane’s prediction, the phenomenon of “cytoprotection” was introduced into the literature by André Robert, who described the unexpected and fascinating finding that prostaglandins, the major metabolic products of arachidonic acid resulting from cyclooxygenase activity, can be crucial for the maintenance of gastroduodenal mucosal integrity. His group provided the experimental evidence that prostaglandins, when applied exogenously in nonantisecretory doses, exhibit high activity in preventing the mucosal damage induced by necrotizing substances, such as ethanol, hyperosmolar solutions, strong acids, alkaline substances, concentrated bile, and even boiling water. The precise mechanism of this so-called cytoprotective property of prostaglandins remained unknown, but the stimulatory effects of these agents on gastric mucus and bicarbonate secretions, an increase in the gastric microcirculation, and the enhancement in the mucosal sulfhydryl compounds were initially proposed to explain this phenomenon.
Prostaglandins were identified as a group of compounds rather than a single substance, and arachidonic acid was identified as their precursor. It was recognized that prostaglandins were produced by nearly all biologic tissues. Profound pharmacologic effects of these compounds were shown on smooth muscle contraction, cell secretions, and platelet aggregation, which fostered the concept that prostaglandins functioned as local mediators of many biologic systems.
The recognition by Schwarz that the formation of gastroduodenal ulcers is caused by the erosive properties of endogenous gastric acid (Schwarz dictum: “no acid-no ulcer”) and that the inhibition of acid secretion enhances ulcer healing, altered the management of this disease from surgery as the mainstay to pharmacologically oriented strategies. Following the discovery of cytoprotective activity of prostaglandins (PGs), stable PG analogs were developed, with the notion that they could play an important role in the pharmacologic treatment of peptic lesions by effectively inhibiting gastric acid secretion in humans. These analogs were found to be effective in accelerating healing ulcer rate not only with associated NSAID therapy, in the presence of an endogenous prostaglandin deficiency, but also in NSAID-independent peptic injury. It was demonstrated that misoprostol (a PGE1 stable analog) significantly decreased the frequency of gastroduodenal ulcers in long-term NSAID users; however, this effect occurred principally as a result of gastric acid inhibition rather than by a cytoprotective mechanism, indicating that cytoprotection plays a lesser role in healing chronic peptic ulcers. Prostaglandins inhibit H+ ion generation by binding to their EP3 G-protein linked receptor on the parietal cell (see Chapter 2, Figure 2.2), which appears to inhibit adenylate cyclase and thereby decrease intracellular cAMP generation when activated.
Physicochemical properties
Misoprostol, racemic methyl (11a, 13E)-11,16-dihydroxy-16-methyl-9-oxoprost-13-en-1-oate C22H38O5 is a water-insoluble, synthetic prostaglandin E1 (PGE1) analog. The commercially available product is a double racemate of two diastereoisomers containing four stereoisomers prepared in a matrix of hydroxyl propyl methyl cellulose.
Formulations and recommended dosages
Misoprostol tablets (Table 4.1) are commercially available under brand names Cytotec® (100 and 200 μg) and Arthrotec® (enteric-coated core that contains either 50 or 75 mg of diclofenac sodium and an outer layer containing 200 μg misoprostol; originally G.D. Searle, Skokie, IL, and now Pfizer, New York, NY). For patients who need protection from NSAID-induced gastroduodenal mucosal injury, the usual adult dosage is 200 μg 3–4 times per day, taken with food. For treatment of duodenal ulcer, 4–8 weeks of therapy with 200 μg 4 times per day is recommended. The recommended dosage of Arthrotec is one tablet 2–4 times per day. With all these regimens, the possibility of abdominal cramps and diarrhea, as well as the risk of spontaneous abortion in pregnant women, should be considered. Although not recommended by the manufacturer, to induce abortion, misoprostol is given with other drugs, for example, misoprostol 400–600 μg after RU-496 600 mg once or in two equal divided doses.
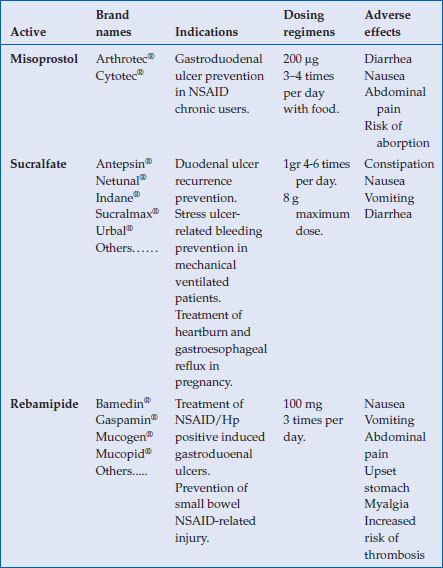
Table 4.1 Data summary of prostaglandins and other mucosal protecting agents
Mechanism of action
The antisecretory properties of misoprostol are produced by only one of the diastereoisomeric pairs. Of the two enantiomers making up that diastereoisomeric pair, the 11R, 16S isomer accounts for most, if not all, of the gastric acid antisecretory activity. The presence or lack of stereospecificity of other pharmacologic activities of misoprostol has not been reported. It is reasonable to expect that some effects, such as those derived from misoprostol competing with PGE1 at its binding site, will show significant stereospecificity.
In addition to its antisecretory properties, misoprostol induces marked edema of the mucosa and submucosa. This mucosal edema and increased mucus layer may represent integral components of the drug’s cytoprotective mechanisms of action. Edema may dilute the concentration of the gastric secretions and increase the distance that damaging molecules, such as NSAIDs, must penetrate before reaching susceptible cells. Misoprostol, as most commonly administered in the clinical setting, has four dominant effects: gastroduodenal cytoprotection, its approved therapeutic indication, and three side effects, namely, diarrhea, abdominal pain, and uterine contractions. For the optimal delivery of drugs that affect the gastrointestinal tract, it is important to determine whether pharmacologic benefits and toxicity are elicited topically on physical contact or systemically to the site of action after absorption. Convincing animal data indicate that misoprostol’s cytoprotective effect occurs principally by topical contact.
Prostaglandins are potent mediators generated in various organs by the enzymatic action of cyclooxygenase on arachidonic acid, and misoprostol, as a prostaglandin analog, might have agonist, antagonist, or both activities relative to endogenous prostaglandins. Misoprostolic acid is an EP2-EP3-selective agonist in intestinal mucosa, preventing the release of a variety of tissue damaging cytokines and inflammatory mediators and by helping to maintain normal homeostasis. Prostaglandins may exert protective gastrointestinal tract effects by inhibiting the release of platelet-activating factor, tumor necrosis factor (TNF), and histamine from mast cells. It is also likely that misoprostol acts as an inhibitor of leukocyte adherence and/or directly modulates the expression of specific adhesion molecules throughout the body in various disease states. Some of the drug’s effects may be due to downregulation of various cytokines. In the presence of lipopolysaccharide (LPS) and human peripheral blood monocytes, 10−6 M misoprostol significantly depressed LPS stimulation of interleukin-1, thromboxane B2, and TNF, while further stimulating the production of 6-keto-prostacyclin. A novel delivery system consists of misoprostol molecules attached to a polybutadiene polymer backbone by hydrolyzable covalent bonds. The formulation was designed to release the drug at a controlled rate, but only in an acidic environment. This formulation allowed the release of potentially active misoprostol from the polymer backbone only in the region of the stomach. In addition, whereas the immediate release product caused the expected frequency of diarrhea in test animals, the side effect was negligible after the polymer at all dosages tested in humans. These results indicate that the cytoprotective effects may require little or no drug reaching the systemic circulation and that diarrhea can be eliminated by the combination of very low systemic availability and no release of misoprostol in the intestinal lumen.
Drug interactions
Many studies have evaluated potential interactions between misoprostol and various NSAIDs (aspirin, diclofenac, ibuprofen, indomethacin, piroxicam); however, no clinically significant interactions have been reported to date. The single-dose pharmacokinetics of misoprostolic acid 200 μg did not change significantly when given alone or with aspirin 975 mg. Similarly, no significant differences in ibuprofen and diclofenac pharmacokinetics were detected when administered with misoprostol. Potential pharmacokinetic interactions with other classes of drugs also were investigated. Misoprostol does not change the pharmacokinetics of antipyrine, suggesting that it does not induce hepatic enzymes. Simultaneous administration of misoprostol 800 μg did not cause significant changes in steady-state diazepam and nordiazepam plasma levels. Misoprostol 400 μg twice daily did not alter single-dose or steady-state pharmacokinetics of propranolol given 80 mg twice/day for 2 weeks. One case report suggested a possible increased prothrombin time after 8 days of treatment with a combination of misoprostol 150 μg per day and diclofenac 400 mg per day.
Pharmacokinetics
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


