CHAPTER 10
Nucleoside analogs
Introduction
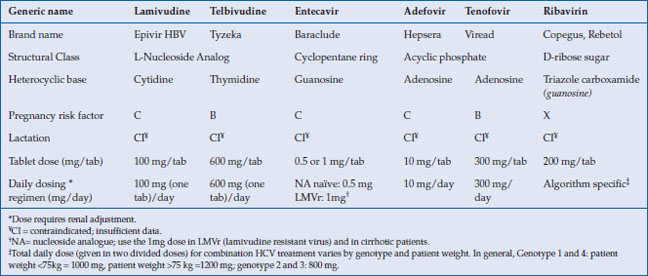
The introduction and approval of acyclovir in 1982 heralded an era of oral antiviral medications that have been used to treat herpesviruses (HSV 1 and 2, VZV), hepadnaviruses (hepatitis B), flaviviruses (hepatitis C), and lentiviruses (HIV), among others. Acyclovir, an acyclic guanine nucleoside analog (NA), is also a prototype of the nucleos(t)ide class of oral antivirals which act by inhibition of viral polymerase or reverse transcriptase. These nucleos(t)ide reverse transcriptase inhibitors (NRTI), some of which were initially used for treatment of HIV, have been found to be effective against hepatitis B. The nucleos(t)ide analogs (NAs) used for the treatment of hepatitis B fall into three structural categories: L-nucleoside analogs (lamivudine, telbivudine and emtricitabine), acyclic phosphonates (adefovir and tenofovir), and cyclopentane rings (entecavir).
The first antiviral agents used for the treatment of hepatitis B were the interferons (see above, Chapter 9). Their use has been limited by their side effects and limited efficacy. However, they have the advantage of a defined course of treatment, absence of viral resistance, and a broad antiviral activity. Furthermore rates of hepatitis B e and surface antigen seroconversion are higher than those achieved with nucleos(t)ide analogs used over a similar duration. Nucleos(t)ide analogs offer a more favorable adverse event profile, oral administration, and unlike interferons, can in most instances be used safely in patients with advanced liver disease. They carry the disadvantages of resistance development and need for long-term viral suppression.
Lamivudine, an L-nucleoside analog, became the first oral medication approved for chronic hepatitis B (CHB) in 1998. While very well tolerated it is no longer a first-line agent because long-term use results in high rates of viral resistance. Adefovir, an acyclic nucleotide analog, which was also first developed for the treatment of HIV, was approved at a lower dose for the treatment of hepatitis B in 2002. Unfortunately, it too suffered from high resistance rates and a risk of kidney toxicity with long-term use. In 2005, entecavir, a guanosine nucleoside analog, was introduced and has demonstrated excellent long-term efficacy with low rates of viral resistance. This was followed in 2008 by the approval of tenofovir, an acyclic phosphonate diester of adenosine monophosphate, which had also been developed for the treatment of HIV and was found to be a potent inhibitor or HBV DNA polymerase. Like entecavir, it is associated with low levels of viral resistance even with long-term use. Both now serve as first-line therapy for CHB. Other nucleoside analogs with activity against hepatitis B include the L-thymidine analog telbivudine and emtricitabine, a cytidine analog similar to lamivudine (Table 10.1).
Table 10.1 Nucleos(t)ides used in hepatitis B and C
Combination treatments with interferon and a NA have not been shown to increase efficacy in the treatment of CHB but this is an area which continues to be investigated. However, combination of ribavirin, a guanosine nucleoside analog, with interferon and a protease inhibitor, has become the cornerstone of chronic hepatitis C treatment. The mechanism of action of ribavirin is incompletely understood and is likely multifaceted. It is hence described separately in this chapter.
Mechanism of action
The nucleos(t)ide analogs enter cells by passive diffusion and possibly a carrier-mediated process. Nucleos(t)ide analogs require phosphorylation by either host cytoplasmic or viral kinases to form the active triphosphate substrate. While the L-nucleoside analogs and cyclopentane rings (a nucleoside analog) require three phosphorylations, the acyclic phosphonates are monophosphates (nucleotide analogs) requiring only two additional phosphates. Potency of the NAs in cell culture assays is partially influenced by the efficiency of the phosphorylation step. For example, entecavir is very efficiently converted to the triphosphate active substrate, a factor thought to be important in defining its greater potency in cell culture assays compared with the other NAs.
The resultant triphosphate active substrates act as competitive inhibitors of the viral DNA polymerase and reverse transcriptase by substituting for the native host nucleotides. Nucleos(t)ide analogs respective affinity to the “Palm” subdomain of the viral reverse transcriptase, the active site of nucleotide incorporation, also determines their potency, as determined by enzymic assays. Since they lack a 3′-hydroxyl group, incorporation of the NA into the elongating proviral DNA leads to chain termination. Though entecavir does have a 3′-hydroxyl moiety on its cyclopentyl group, it nevertheless halts elongation after a few nucleotide additions, hence still serving as a de facto chain terminator. While all the above-mentioned NAs obstruct the reverse transcription of pregenomic mRNA to the negative viral DNA strand as well as the next step of positive strand synthesis, entecavir also impedes the initial step of base priming by the polymerase. Though NAs slow or halt viral replication, they do not eradicate infected cells that already have proviral DNA integrated into their genome or prevent infection of susceptible cells by existing virus. Therefore, notwithstanding the rare event of a hepatitis B surface antigen (sAg) seroconversion by the host, they do not lead to a cure and have modest effects on HBsAg and eAg expression.
Nucleos(t)ide analogs discussed here have a very low affinity for human DNA polymerases including DNA polymerase-α and –β and mitochondrial DNA polymerase γ. Some NAs used in combination HIV therapy have a greater affinity to human mitochondrial DNA polymerase-γ and can lead to serious adverse events including lactic acidosis, pancytopenia, myopathy, peripheral neuropathy and pancreatitis. These side-effects are rarely seen with the NAs used to treat HBV infection as these agents have very low affinity for the human DNA polymerases and these effects are less common when used as monotherapy. An increased incidence of these complications has been shown to occur in HIV positive patients on antiretroviral therapy receiving ribavirin and interferon for HCV infection. A few cases of entecavir associated lactic acidosis have also been reported in patients with advanced liver disease.
Since NAs interfere with DNA replication there has been some concern that they may have an oncogenic effect. Entecavir has been associated with an increased risk of lung tumors in mice at doses 3–5 times greater than those use in humans. However this effect appears to be species specific. Doses 30–40 times the human equivalent have been shown to cause lung, brain and liver tumors in mice and rats.
Drug resistance
Mutations in the viral genome altering the nucleotide binding site on the reverse transcriptase confer drug resistance. The M204V substitution on the YMDD loop of the “palm” subdomain leads to lamivudine resistance by hampering this loop’s anchoring of the triphosphate of the nucleotide and changing the pocket shape of the nucleotide binding site causing steric hindrance. This mutation is often accompanied by a compensatory adaptive mutation, L180M, restoring some viral replication fitness. It has full cross-resistance to the other structurally similar L-nucleoside analogs such as telbivudine and emtricitabine. The M204V + L180M substitutions lead to partial cross-resistance with the structurally different entecavir. An additional substitution at T184, S202, or M250 that further restricts the active site pocket, is needed for full entecavir resistance. Hence complete entecavir resistance requires multiple viral mutations, explaining in part, why entecavir has such a high barrier to resistance. The A181V and N236T substitutions that lead to adefovir and tenofovir resistance alter how nucleotides fit into the active site pocket more indirectly by disrupting secondary stabilizing interactions.
Pharmacology
The NAs ease of use is attributable to some of their favorable pharmacological properties including generally high oral bioavailability, efficient cellular passive and active transport, longer cellular half-life of the active substrates, absence of cytochrome P450 metabolism, and renal excretion of unaltered drug (Table 10.2). The L-nucleosides and cyclopentane rings have high oral bioavailability, characteristically over 80%. The greater polarity of the nucleotide analogs due to their acyclic phosphonate group leads to poor oral bioavailability. This is circumvented by the development of diester prodrugs. Upon absorption, adefovir dipivoxil and tenofovir disoproxil are readily hydrolyzed by plasma and intestinal epithelium esterases to the active drug. In the case of adefovir, the liberation of the pivalic group contributes to the toxicity profile at higher doses.
Table 10.2 Pharmacokinetic properties of NAs
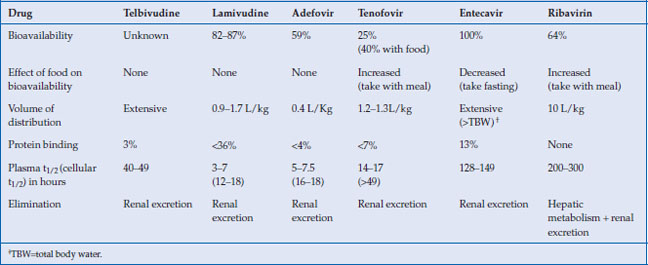
The NAs generally have a volume of distribution equal to or exceeding total body water. They are scantly protein bound. Due to the efficient renal excretion of the parent drug, the plasma half-lives of the NAs discussed here are short, with the notable exception of entecavir. However, the intracellular phosphorylated active drug species have a longer half-life and consequently allow for once daily dosing.
The NAs do not undergo catabolism but are largely excreted unchanged by the kidney. As a result they are associated with only few drug–drug interactions. Furthermore, NAs should be dose adjusted in renal but not liver failure. Renal excretion occurs through both glomerular filtration and tubular excretion. The acyclic phosphonates (adefovir and tenofovir) undergo efficient transport at the renal proximal tubules by organic anion transporter (hOAT) and their accumulation there explains the associated renal toxicity. In addition, there may be competition for excretion with other drug substrates of hOAT, such as some protease inhibitors, resulting in drug–drug interaction.
Clinical effectiveness
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


