CHAPTER 3
Histamine H2-receptor antagonists
Introduction
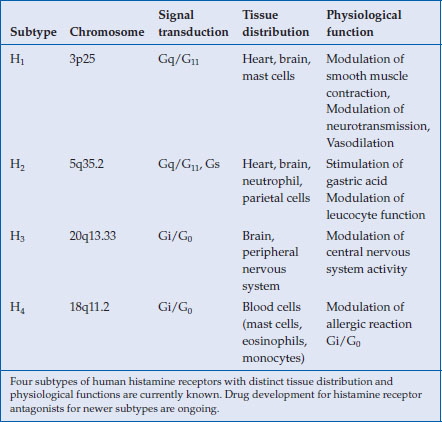
Drug development targeting human histamine receptors have been rewarded with the successful launching of H1– and H2-receptor antagonists in clinical use. Currently, two additional histamine receptors (H3 and H4) have been identified, for which intensive efforts have been made for developing new drugs (Table 3.1). These new antihistamine H3– and H4-receptor antagonists are aimed at treating allergic diseases (asthma, allergic rhinitis) or disorders affecting the central nervous system (Alzheimer’s disease, narcolepsy) and are still in their clinical development stages. Because their application in digestive diseases remains uncertain, this chapter will focus on H2-receptor antagonists and their clinical pharmacology.
Table 3.1 Human histamine receptors
Mechanism of action
Histamine has been recognized as a potent stimulant of acid secretion, but conventional antihistamine (H1) compounds used to treat allergic conditions generally possessed poor inhibitory capacity in terms of acid suppression. Therefore, the presence of a separate class of histamine receptor responsible for acid secretion was predicted and, as a result, intensive efforts for drug development were pursued. In the early 1970s metiamide, the first compound specific to the H2 receptor, was evaluated for the treatment of peptic ulcer diseases and demonstrated superior effects over a placebo in terms of symptomatic relief and ulcer healing. Unfortunately, metiamide was withdrawn early due to bone marrow toxicity, which occurred as a result of its thiourea group. Subsequently, however, with the replacement of the thiourea for a cyanoguanidine group, cimetidine was introduced into clinical use. It revolutionized the management of peptic ulcer diseases with dramatically improved effects on symptomatic control, as well as ulcer healing, compared to conventional treatment with antacids and anticholinergic drugs.
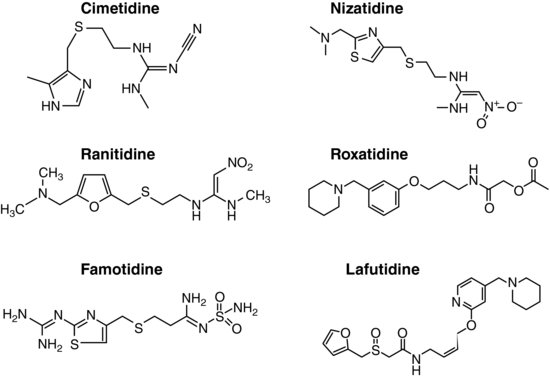
Following the brilliant success of cimetidine, many drugs acting as H2-receptor specific antagonists (H2RAs), such as ranitidine, famotidine, and nizatidine, have been developed and marketed all over the world. Two more H2RAs, roxatidine acetate and lafutidine, are also available in Japan and other Asian countries (Figure 3.1).
These compounds bind to histamine H2 receptors on parietal cells (see Chapter 2, Figure 2.2) and thereby antagonize the action of histamine. In addition, some of these H2RAs have other actions unique to each compound, such as immune modulation by cimetidine, stimulation of salivary secretion by nizatidine, and stimulation of gastric mucus secretion by roxatidine and lafutidine.
Although histamine is the major activation signal for stimulating acid secretion, other secretagogues, such as cholinergic stimulation or gastrin, can also stimulate acid secretion independent from histamine. Therefore, acid inhibition by H2RAs is less potent compared to proton pump inhibitors (PPIs) that target H+, K+-ATPase, which is the final common enzyme responsible for acid secretion activated by all secretagogues.
Pharmacology
Most H2RAs developed early have imidazole (cimetidine), furan (ranitidine), or thiazole (famotidine and nizatidine) structures that simulate the imidazole ring of histamine as their chemical backbones. Newly introduced H2RAs, such as roxatidine acetate and lafutidine, however, have different chemical structures (Figure 3.1). All of these drugs are administered orally, but parenteral preparations are also available for most of these drugs. The absorption of H2RAs is rapid and reaches peak concentration in 1–3 hours. They all show good bioavailability, ranging from 30 to 100%.
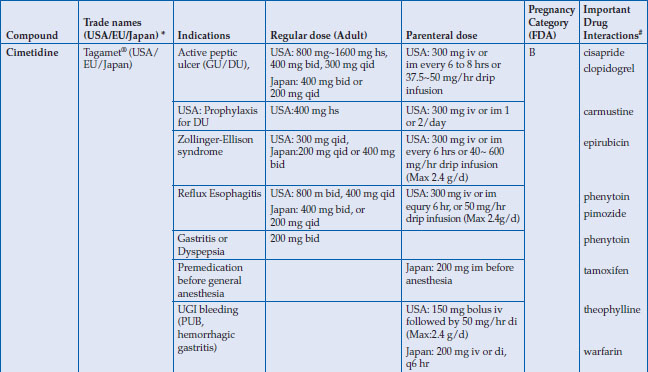
Regular doses for healing peptic ulcers are quite different (Table 3.2), which reflects their relative potencies with regard to acid inhibition. Although once daily regimens are effective in healing ulcers, especially when given at bedtime, they are commonly given in two divided doses because of their short serum half-lives (1–4 hours). In general, H2RAs are metabolized in the liver, but renal excretion also contributes to their elimination. There are considerable differences in the relative contributions of hepatic and renal pathway for each drug. For nizatidine, the kidney plays a major role in elimination, whereas heaptic metabolism contributes more to the disposition of cimetidine, ranitidine, and famotidine. Notably, cimetidine is mostly metabolized through cytochrome enzymes, including CYP2C9 and CYP3A4, which are responsible for metabolizing many other drugs, such as warfarin, and consideration should thus be given when co-prescribing drugs that share the same metabolic disposition pathways. Nevertheless, all these H2RAs require dose reductions when renal function is impaired. The only exception is lafutidine, which undergoes hepatic metabolism, with the majority of the metabolites being excreted into stool.
Table 3.2 Histamine H2-receptor antagonists
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


