Fig. 13.1
Cell cycle (Reprinted from Kalmadi and Raghavan [43]. With permission from Springer Verlag)
1.
Cells remain quiescent in a resting phase (G0), which may be prolonged over years;
2.
Cells that are committed to replication enter the interphase (G1) that is characterized by synthesis of RNA and protein, preparing the cell to enter the next phase;
3.
DNA synthetic (S) phase, in which the DNA content is doubled.
4.
Second resting (G2) phase prior to the cell undergoing mitosis
5.
Mitotic phase (M), in which the chromosomes separate and divide, forming two daughter cells
Mitosis results in cells which consist of (a) non-dividing, terminally differentiated cells; (b) resting cells (G0) which can be recruited into the cell cycle; and (c) continually dividing cells which enter into G1 phase again. Major checkpoints in the cell proliferation occur in G1 when cells must commit themselves to division and in G2 before undergoing mitosis. The cell cycle is under the control of numerous regulatory mechanisms, including cyclins (proteins that are positive regulators) and cyclin-dependent kinases (CDK). CDKs are present in all phases of the cell cycle and control the cascade of proliferative signals. The regulation of CDKs by the cyclin molecules causes their levels to fluctuate, leading to synchronization of the processes of cell division [3].
Chemotherapy and radiation are most effective in achieving cytotoxicity when the cells are in cycle. One approach to the categorization of cytotoxics is based on their activities relative to the cell cycle:
A.
“Phase specific” drugs are effective only if present in the tumor cell during a specific phase of the cell division. Although an increase in the drug level or dose will not result in more tumor kill, exposure over a longer period of time may allow more cells to enter the specific lethal phase of the cycle, thus producing a greater level of cell kill. Examples of this process include the antimetabolites during the S phase, and taxanes and vinca alkaloids during G2 and M phases.
B.
“Phase non-specific” agents have actions not restricted to a specific step in the cell cycle; these can be further divided into (a) cycle nonspecific drugs which can kill non-dividing cells (e.g. steroids, some antitumor antibiotics); and (b) cycle specific agents which can kill cells which enter into the cell cycle (e.g. alkylating agents). Phase nonspecific agents have a linear dose-response curve: the higher the dose administered, the greater the fraction of cells killed.
With improved understanding of the human genome, we have developed more knowledge of the level of molecular checks and balances involved in the regulation of the cell cycle. A detailed discussion of this topic is beyond the scope of this overview, apart from a brief focus that relates to targeted therapeutics (see below).
Tumor Kinetic Modeling
The growth of a tumor can be simplistically conceptualized as being dependent on several variables: the rate of cell loss, growth fraction (proportion of cells in proliferative phase) and cell doubling time. Several models have been devised to explain the impact of therapeutic regimens on the tumor cell cycle [4].
Skipper et al. originally proposed the log kill model, which was based on the behavior of L1210 leukemia cell lines in rodents [5]. They suggested that the benefit of chemotherapy was due to the cytocidal effects on the cancer cells and that tumor growth and tumor regression in response to chemotherapy were exponential in nature. This model proposed that a drug that would cause a reduction of tumor burden from 1012 to 1011, if given in the same dose, would also decrease the burden from 106 to 105 (i.e. each reduction reflected a 90 % tumor cell kill). This formed the basis for the use of repeated cycles of chemotherapy to achieve maximal tumor eradication. However, it has become clear that exposure to the same regimen for more than 4–6 doses does not improve outcomes. We now understand the growth models of tumor have also a mixed nature with respect to different cancer cells in vivo [6]. The Gompertzian sigmoid growth curve is characteristic of many solid tumors. Tumors grow most rapidly at smaller sizes and then the growth rate slows, secondary to problems with vascularity, hypoxia and interaction with the other cells in their microenvironment [7–11].
Concepts initially derived from resistance in antibacterial therapy were applied to the study of antimetabolites in the treatment of murine cancer models and cell lines, and it was shown that resistance is acquired at various points during the growth of the tumor. This also occurs in human tumors. Goldie and Coldman suggested that larger tumors have more cells and thus have a greater chance of undergoing spontaneous mutation [12], which may, in turn, confer resistance to chemotherapy. It has thus been suggested that tumors are more effectively treated at a smaller size, before they have the ability to develop mutations and resistance. This has also led to the concept that non-cross resistant multiple agent chemotherapy would have a greater proportionate tumor kill and could thus prevent the development of resistance.
Another fundamental concept of cell cycle kinetics is the so-called Norton-Simon regression hypothesis [13]. Their concept is that administration of chemotherapy at shorter time intervals (with greater dose-intensity) achieves greater proportionate tumor cell kill. As noted above (Fig. 13.2), some tumors do not grow in a simple exponential manner but rather follow a Gompterzian growth pattern [11, 13]. Norton and Simon have also proposed that less time available for tumor regrowth between treatments will improve the chance of cure, and that sequential, dose-dense, non-cross resistant regimens will minimize drug resistance. This approach destroys the dominant tumor population initially via the first component of treatment, with different agents addressing the residual resistant cells, without the potential for significant tumor regrowth.
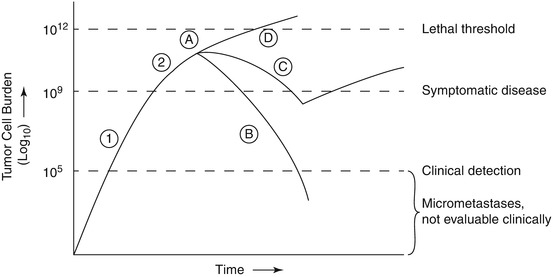
Fig. 13.2
Gompertizan model of tumor growth: Relation between tumor mass, diagnosis, and treatment regimens. Growth phases (1) Exponential growth (2) Plateau phase with slower growth due to cells outgrowing vascular supply and nutritional resources, and possible other regulatory mechanisms. Treatment responses (A) Initiation of treatment; (B) Curative therapy; (C) initial response with secondary resistance; (D) primary resistance (Reprinted from Kalmadi and Raghavan [43]. With permission from Springer Verlag)
The Goldie-Coldman and Norton-Simon hypotheses have also been applied to the design of adjuvant chemotherapy regimens, attempting to rationalize the prior empirical approach. This has resulted in improved cure rates, perhaps due to increased cytotoxicity and/or a reduced emergence of drug resistance [13–15].
Pharmacology of Antineoplastic Agents
The clinical pharmacology of antineoplastic agents has been an evolving field for the past century, with the most progress having been made in the past decade through the advent of analytical tools such as liquid chromatography and mass spectroscopy. Most cytotoxic agents offer a very narrow therapeutic index compared with most drugs [16].
Pharmacokinetic Principles
Pharmacokinetics involves the study of the fate of administered medications, including absorption, distribution, metabolism and excretion. Absorption has historically not been a major issue for oncologists, because for decades most drugs were administered intravenously. With the advances in drug delivery involving orally administered targeted agents and prodrugs, this has become an issue with oral and cutaneously absorbed drugs. The level of bioavailability is calculated by comparing the area under the curve (AUC) of an oral drug to the same dose given intravenously.
Clearance of the drug is the most critical aspect and can be conceptualized as being a function of drug distribution, metabolism and elimination. Although drug distribution can be viewed most simply as being two-compartmental (extracellular and intracellular), it is actually more complex. It should more properly by considered as a multi-compartment model with frequent redistribution within. The presence of sanctuary sites with poor penetration of the drug may result in apparent resistance of tumors that would otherwise be responsive to chemotherapy.
Distribution can also be affected by disease states such as cardiac failure, or by the impact of advanced cancer (e.g. with associated hypoalbuminemia), resulting in pleural effusions and ascites, thus creating pathological compartments.
When third spaces are present, such as pleural effusions or ascites, some agents (e.g. methotrexate) can cause excessive toxicity due to prolonged exposure, since methotrexate accumulates in these third spaces. Elimination is studied along two different models: (1) The linear kinetic model is based on the assumption that the half life of the drug will be constant. (2) Nonlinear kinetics assumes that the elimination of the drug is saturable resulting in different rates of excretion at different concentrations resulting in variable half-lives.
The metabolism of these agents is determined by the amount of resemblance that they show to physiologic substrate. Drugs (eg. purine and pyrimidine analogues) that are similar to normal metabolites are processed by the same mechanisms as the normal metabolites intracellularly. Those that differ from normal metabolites are degraded in the liver, in reactions involving oxidation (controlled by the cytochrome P450 enzymes) and conjugation. The ultimate excretion of the drug from the body is via the hepatobiliary system and/or the kidney. Creatinine clearance, often used as a surrogate marker of renal function, assesses the glomerular function rate. This index is subject to the influence of muscle mass, and may be misleading in the elderly patient with reduced muscle bulk. Tubular secretion and reabsorption may also play a role in drug excretion. Changes in the renal excretion of the drugs can alter their efficacy and toxicity profile.
Hepatic excretion involves a number of transport systems including P-glycoprotein and canalicular multispecific organic anion transporter (cMOAT). Enterohepatic recirculation also plays a major role, especially for the drugs metabolized by the glucuronide pathway. Changes in the regulation of the cytochrome system by other medications being used may cause changes in the bioavailable levels of chemotherapeutic agents metabolised by this system. The use of liver function tests to modify the dosage of these drugs is thus fraught by numerous pitfalls, since it does not always accurately predict the toxicity risk due to the fact that they do not accurately estimate the level of dysfunction [17].
Another important component of the system is inter-patient pharmacokinetic variability [18]. Pharmacogenetic variation explains some of the variability in the response and toxicity of patient groups to agents like 5-FU and irinotecan [18, 19].
Deficiency of dihydropyrimidine dehydrogenase, reflecting different polymorphisms of the DPD gene, which inactivates 5-FU, will lead to increased toxicity from this cytotoxic. The active form of irinotecan, SN38, is inactivated via glucuronidation. Reduced activity of UGT1A1, which is involved in this glucuronidation, and which is a sine qua non of Gilbert’s syndrome, leads to dose limiting diarrhea and neutropenia.
Other factors may also cause inter-patient variation in pharmacokinetics. The variability in absorption of oral drugs secondary to chemotherapy induced damage to the mucosa can alter efficacy and toxicity of these agents. Most chemotherapy regimens are dosed based on body surface area, which is calculated by using body weight and height. Obesity, which causes increase in the lipophilic compartment, is not well addressed by this calculation of drug dosing. The amount of lipid solubility of a drug can cause changes in the drug levels in the obese. Hypoalbuminemia causing decreased binding and increased free concentration of the drug can increase side effects. This may be quite important in a pretreated patient with advanced cancer and cachexia, which is commonly associated with hypoalbuminemia.
Pharmacodynamics
The fundamental objective of pharmacodynamics is to understand dose-response relationships, usually predicated on the assessment of drug levels in different tissues [19, 20], as well as the pharmacogenomic considerations noted above. Initial pharmacodynamic principles were based on the concept that all drugs will have a sigmoidal shape in their drug effect, based on the theory that drugs require a receptor interaction for their effect. However, this rule is not valid for phase specific antineoplastic agents. When cells are not in the specific phase, increasing the dose will not increase the sensitivity, although increasing the time of exposure may have the desired effect.
The prediction of toxicity and response in an individual patient should be based on both pharmacokinetic and pharmacodynamic principles. Reducing drug dose for excessive toxicities may seem to be logical in the case of altered excretory function, but may not be effective in the case of reduced hematological reserve after prior treatment, resulting simply in under-dosing.
Monitoring of drugs with a narrow therapeutic index is usually done by monitoring drug levels in the body, in most other areas of medicine. This is more complicated in cancer treatment because of the frequent use of combination chemotherapy. Techniques such as low-dose weekly scheduling (e.g. taxanes) and AUC dosing based on creatinine clearance (e.g. carboplatin) are being used to limit toxicity without compromising efficacy.
Patterns of Drug Response and Resistance
Response of tumors can be divided into three broad groups [21, 22]: (a) drug sensitive tumors, with treatment resulting in cure; (b) highly responsive tumors, but with eventual refractoriness to cytotoxic treatment; and (c) tumors with little responsiveness to chemotherapy (Table 13.1).
Table 13.1
Responsiveness of genitourinary tumors to chemotherapy
1. Highly sensitive |
A. Childhood cancers, such as Wilms tumor, Ewings tumor, and rhabdomyosarcoma |
B. Germ cell tumors – e.g. of testis, retroperitoneum |
C. Lymphoma (extranodal) – Hodgkin’s disease; Burkitt’s lymphoma |
2. Moderately sensitive |
A. Adenocarcinoma of the prostate |
B. Transitional cell carcinoma of bladder and urothelial tract |
C. Squamous cell carcinoma of penis or urethra |
D. Some non-Hodgkin’s lymphomas involving genitourinary tract |
E. Small cell anaplastic carcinoma – prostate, bladder |
F. Sarcomas of bladder or prostate in adults |
3. Less/Minimally sensitive |
A. Adrenal gland cancers |
B. Renal carcinomaa |
C. Adenocarcinoma of bladder |
Drug resistance has been studied in both in vivo and in vitro in a range of models. Multiple reasons involving anatomical, pharmacological and biochemical mechanisms explain various aspects of tumor resistance to cytotoxic chemotherapy:
Reduced intracellular levels secondary to transport system inhibition (e.g. the folate transport mechanism leading to methotrexate resistance), reduced diffusion across the cell membrane or increased efflux (P-glycoprotein MDR1 drug efflux pump). Classical multidrug resistance (MDR) is associated with the over-expression of P-glycoprotein (MDR1, P-170) [23, 24]. This causes increased efflux of various antineoplastic agents from the cell leading to decreased accumulation intracellularly. This has been implicated in the cross-resistance patterns between anthracyclines (e.g. doxorubicin), taxanes (e.g. paclitaxel), vinca alkaloids (e.g. vincristine) and the topoisomerase inhibitors (e.g. etoposide). Tumors, which over-express this gene, have sometimes demonstrated increased resistance and poor response to chemotherapy;
Defects in cellular death mechanisms. Alkylating agents cause cell death by intra-strand DNA linkages. This results in defective cell repair systems that should lead to cell death. However, if this system does not recognize the DNA defects, this will prevent tumor cell death, resulting in resistance [15, 25]. Defects in the apoptotic pathway can also involve the Bcl-2 family of proteins and other regulatory mechanisms, such as p-53. Bcl-2 family proteins include both up- and down- regulators of apoptosis. Chemotherapy induced damage to cells is perceived by p53 which can then initiate either apoptosis or cell repair. Altered p53 is one of the most common genetic abnormalities seen in solid tumors. Expression of wild type p53 and changes in Bcl-2 family members can result in altered sensitivity of the tumor cell to chemotherapy agents [26, 27];
Alteration of drug targets, including receptors or enzymes (e.g. thymidylate synthetase in 5-FU resistance) [27];
increased levels of cell protective agents (e.g. glutathione in cisplatin resistance), which prevent oxidative damage and death of the cell via cell scavenging functions, have been implicated in preclinical models of bladder cancer [28];
Modification of drug metabolism can be catastrophic to antineoplastic agents that are designed as prodrugs (e.g. cyclophosphamide must be activated in the liver; irinotecan must be converted to SN-38, its active moiety);
Approaches to circumvent drug resistance have involved the use of multidrug combinations, dose escalation, agents that reverse increased efflux, cofactors that amplify drug efficacy, and inhibition of drug inactivation. Recently liposomal and nanoparticle albumin- stabilized formulations have been applied to overcome drug resistance, but with limited success in genitourinary malignancy [31]. These increase the delivery of chemotherapy to the tumor cell while minimizing toxicity.
Combination Chemotherapy Principles
Single agent regimens, with few exceptions (e.g. gestational choriocarcinoma), are rarely able to achieve cure. In view of this and the observations above, combination chemotherapy regimens have been devised to accomplish the major objectives of attaining maximum tumor kill with minimal toxicity and to prevent drug resistance [16, 32]. The era began when numerous active drugs became available simultaneously and were applied to curative management of leukemias and lymphomas in the late 1970s. Fundamental principles used in the selection of the drugs for combination regimens include [32]:
Use of drugs with defined activity against the tumor
Known expression of different patterns of resistance
Different mechanisms of action with potential synergy
Non overlapping dose-limiting toxicities
Optimal dosing and timing in combination to allow the shortest treatment-free interval
Absence of negative interactions – e.g. adverse interactions between cytotoxics and targeted therapies, negative summation effects between two cytotoxics
The relationship between doses and combination of these agents is complex [32, 33]. The maintenance of dose intensity has proved to be important in the success of some of these regimens. Reduction of dose can result in significantly decreased cure rates, especially in the more responsive tumors such as germ cell cancer. Thus, although responses may occur despite dose reduction, residual tumor cells often persist, leading to eventual relapse and decreased survival. The concept of relative dose intensity (amount of drug delivered in a given time frame) has evolved over the past two decades, but it remains controversial as to whether this is a major factor in cure [33].
Ideally drugs should be used in their optimal schedule and dosage even when being combined with other agents. However, care must be exercised to avoid over-dosage and excessive normal-tissue toxicity. In general, the interval of drug delivery also needs to be consistent, keeping the treatment free interval to be the shortest time necessary for resolution of any dose limiting toxicity (usually bone marrow toxicity) to maintain dose intensity [32–34].
The selection of patients to receive combination chemotherapy has also undergone refinement. In the present era, we are more cautious than in the past, and many oncologists believe that it may not be appropriate for a patient with a poor performance score (e.g. ECOG level 3–4) to be given a toxic regimen, unless there is substantial evidence that his disease is likely to be highly responsive to the treatment [35]. We believe that it is critically important to discuss the aims and expectations of chemotherapy with all patients, but of even greater importance in the elderly and infirm. By contrast, with better understanding of age-related changes in drug pharmacology and the importance of performance status, more venturesome approaches are now being explored in the chemotherapy of the fit elderly with advanced cancer [36].
High Dose Chemotherapy and Stem Cell Transplantation
High dose chemotherapy involves the use of potentially lethal doses of chemotherapy with or without radiation, followed by rescue with hematopoietic stem cells obtained from the blood or by marrow harvest and storage [37–40]. This is predicated on the concepts that (a) there is a dose response relationship for a specific regimen in certain tumors; (b) there is benefit to dosing at a higher level than can be survived by normal tissues (e.g. bone marrow), provided that re-infusion of bone marrow can avoid the death of the host. This modality, which is predominantly used in hematological malignancies, has been applied to the management of relapsed or poor-risk metastatic germ cell tumors [38]. To date, despite evidence of anti-cancer activity, prospective, randomized trial data have not shown an unequivocal survival benefit from this approach to the management of advanced germ cell tumors, compared to standard dose poor-risk or salvage regimens [38, 41]. There are still advocates of this procedure in the management of poor-risk or relapsed germ cell tumors, but the majority of evidence to support this contention comes from retrospective, historically controlled analyses [42].
Allogeneic Bone Marrow Transplantation
This process involves the acquisition of stem cells from a donor who has complete/partial HLA-compatibility with the patient [39, 40]. This can include matched related donor, matched unrelated donor (e.g. HLA matched donor from the Bone marrow registry), stored cord blood, syngeneic (identical twin), and haplo-identical transplantation (e.g. sibling/parent who is half matched to the patient). Complexities of allogeneic bone marrow transplantation involve immunosuppression after the transplant to prevent rejection of the donor cells by the host. This milieu of intense cytotoxic damage to the bone marrow and immunosuppression allows the donor graft cells to launch a response against the recipient termed as graft versus host disease (GVHD). GVHD can also have a positive effect on the tumor by having a graft versus tumor effect; which can be curative in some malignancies like Chronic Myelogenous Leukemia. Advantages of allogeneic bone marrow transplantation include the graft versus tumor effect, curative option in patients with tumor involvement of the bone marrow and no tumor contamination of the graft cells.
Disadvantages include graft versus host disease; higher acute treatment related mortality, higher infectious complications secondary to immunosuppression needed after transplant and the need to locate a suitable donor. In addition, increasing evidence is available regarding late complications, including the development of second malignancies [40].
Autologous Bone Marrow Transplantation
This variant of marrow transplantation uses the patient’s own hematopoietic stem cells, which are harvested and cryopreserved prior to initiation of treatment [38]. After the completion of high dose chemotherapy and/or radiation the harvested marrow is reinfused. Advantages include the absence of requirement for immunosuppression after infusion of stem cells, no Graft Versus Host Disease, the potential for use in older patients; no requirement for a donor; and lower treatment related mortality of about 2–5 %.
Some of the disadvantages of this approach include the absence of graft versus tumor effect, and the risk of re-seeding tumor cells if there is marrow involvement by the malignancy.
Brief Overview of Cytotoxic Agents
Alkylating Agents
The alkylating agents are among the oldest cytotoxic agents, and form the backbone of numerous regimens. They impair cell function by transferring alkyl groups to amino, carboxyl, phosphate or sulfhydryl groups of nucleic acids (DNA & RNA). The most actively alkylated site is the N-7 position of guanine. This results in cross-linked DNA strands that cannot replicate, impaired transcription of RNA and other damage to the genetic material. They are cell cycle specific, but not phase specific. They have traditionally been divided into five classes, although the platinum complexes (with similar, bifunctional alkylating action) have been included as a sixth class.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


