INITIAL PATIENT ASSESSMENT
Patient assessment begins with a complete past medical history and physical examination. Taking a thorough past medication history, drug allergy, adverse drug reactions, and concurrent medicines are vital in the initial evaluation of patients with chronic kidney disease. Patients with kidney disease receive on average 11 different medications for the treatment of cardiovascular diseases, metabolic disorders, infectious complications, and other comorbid conditions. In addition, patients with kidney disease experience a threefold higher incidence of adverse drug reactions compared to the general population with normal renal function (7–9). Pharmacotherapy should be individualized and minimized to reduce the risk of adverse drug reactions and potential drug–drug interactions.
Principles of Altered Pharmacokinetics in Kidney Disease
Several pharmacokinetic factors are altered when renal function is impaired. These factors include bioavailability, volume of distribution, protein binding, and biotransformation. Pharmacologic agents may pass through several body compartments before reaching receptor site or being eliminated from the body (FIGURE 35.1).

Bioavailability
The bioavailability of a drug refers to the fraction (expressed as a percentage) of a given dose that reaches the systemic circulation. The rate and route of administration are the primary factors which determine bioavailability. An agent that is not completely absorbed or is eliminated by the liver in its first pass has low bioavailability. Drugs administered intravenously are generally 100% bioavailable because the entire dosage reaches the systemic circulation. When given orally, subcutaneously, or intramuscularly, bioavailability decreases. For example, given intravenously, furosemide is 100% bioavailable but only approximately 50% bioavailable when given orally. In patients with right-sided heart failure or liver disease, the absorption rate may drop to 10%. For this reason, many clinicians double or triple the furosemide dose when switching a patient from IV to oral furosemide (10).
Drug absorption determines bioavailability and may be altered in patients with renal insufficiency. Many factors may account for this phenomenon. Neuropathic changes resulting from diabetes mellitus and/or aging may cause gastroparesis or uremia-induced vomiting. Patients with cirrhosis, nephrosis, or congestive heart failure (CHF) commonly have edematous gastrointestinal mucosa, causing a reduction in drug absorption. Drugs which increase gastric pH such as phosphate binders and H2 receptor blockers may impair absorption of concomitantly administered drugs. Concomitant administration of some drugs (such as aluminum- or calcium-containing phosphate binders) with antibiotics or iron-containing supplements may result in the formation of insoluble complexes which can both decrease absorption and slow gut motility.
Volume of Distribution
The volume of distribution (Vd) of a pharmacologic agent is derived by dividing the total amount of drug in the body by the concentration of the drug in the blood. The Vd does not refer to a specific anatomic compartment, but it is useful for calculating the dose required to achieve a desired systemic drug level. An inverse correlation exists between the serum concentration and the Vd (11,12).
After a drug reaches the systemic circulation or is completely absorbed, it is distributed throughout the body. Initially, drugs are distributed to high-flow organs such as the heart, liver, kidney, and brain. In the next phase, drugs are distributed to areas with a lower extraction ratio such as fat, bone, and skin. The rate and extent of distribution of a drug throughout the body determine how much of the drug will be available to exert the pharmacologic actions and how rapidly the drug will be eliminated. Kidney disease affects both drug distribution and protein binding. Edema and ascites increase the distribution volume of many drugs, whereas volume depletion contracts this space. Fluid-volume status of patients should be assessed before administering drugs that require dose adjustment for body weight. For most drugs, ideal body weight (IBW) should be calculated. For men, IBW is 50 kg plus 2.3 kg for each inches more than 5 ft. For women, IBW is 45.5 kg plus 2.3 kg for each inches more than 5 ft.
Therefore, alterations in the extracellular fluid volume can affect the Vd. Volume contraction decreases the Vd while serum drug concentration rises. This is particularly true for hydrophilic compounds such as aminoglycosides or drugs with low Vd (less than 0.7 mL/kg). A rise in extracellular fluid volume due to edema or ascites increases the Vd, resulting in a lower serum drug concentration.
Protein Binding
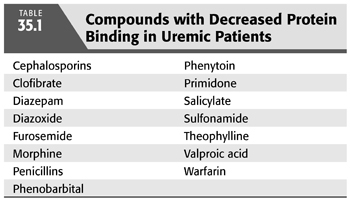
Plasma contains many proteins which bind drugs. Plasma protein binding is often altered in kidney disease (TABLE 35.1). Most drug molecules are circulating in both bound and unbound (free) forms. It is the unbound drug which is distributed and is biologically active (13). The concentration of a given agent which is bound to plasma proteins may be considered a storage pool for that agent. The effects of diseases on drug–protein binding are complex; however, kidney disease tends to decrease protein binding for most agents and increases the free fraction. This is, in part, true because circulating organic wastes bind to carrier proteins, thereby displacing the pharmacologic agent. As a result, a larger concentration of the agent circulates in its free, active form. Most drug assays measure total drug concentration that contains both bound and free drug levels. In some cases (such as in a patient receiving phenytoin), it may be prudent to specifically monitor unbound drug when a narrow therapeutic window exists.
Biotransformation
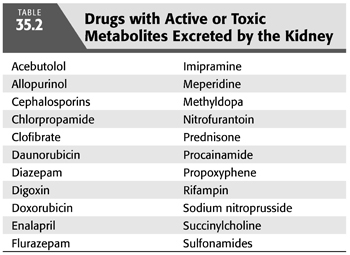
Metabolism or biotransformation refers to the biochemical conversion of a drug from one chemical form to another (10,14). Most biotransformation occurs in the liver through hepatic metabolic pathways including oxidation, reduction, acetylation, or hydrolysis. The result is a more polar, hydrophilic metabolite which is more readily excreted. Many drug metabolites are pharmacologically active and depend on renal excretion for elimination from the body. Kidney disease affects both renal and nonrenal pathways. There is some clinical evidence of slow hydrolysis and faster oxidation rates in patients with kidney disease. Toxic metabolites dependent upon renal excretion may also form (TABLE 35.2).
Elimination
The glomerular filtration rate (GFR) is closely correlated with renal drug elimination and is useful in determining dosage adjustments. If the total renal clearance of a drug or active metabolite is greater than 30%, a dosage adjustment is needed.
To obtain metabolic balance in patients with reduced renal function, several agents should be avoided. Some drugs increase the metabolic load by increasing creatinine production and/or urea production (glucocorticoids and androgens). Other drugs may overwhelm the kidney’s capacity by increasing the metabolic load of excess acid, alkali magnesium, potassium, or sodium. Nonsteroidal anti-inflammatory agents may impair the kidney’s ability to excrete free water.
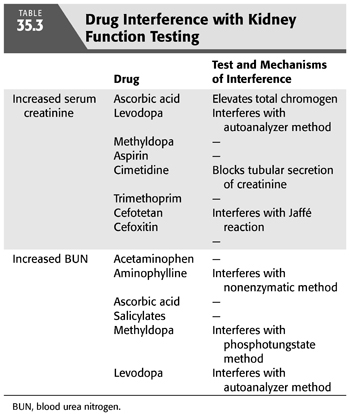
Various pharmacologic agents can interfere with laboratory markers of renal function (TABLE 35.3). Elevated serum creatinine (Scr) from direct drug interference with the chromogen assay or by inhibition of renal tubular creatinine secretion can cause confusion to the inexperienced clinician. Similarly, a number of agents can also cause a rise in the blood urea nitrogen (BUN) through interference with the assay utilized. Temporary discontinuation of the offending medication may be necessary when the Scr or BUN rises unexpectedly. The Cockcroft-Gault (CG) formula is widely used to calculate the creatinine clearance (Clcr) and has traditionally been used to approximate the GFR (15). BUN and Scr are, at best, crude markers of kidney function. The CG formula includes the variables of age (years), IBW (kg), and Scr (mg/dL) and calculates the Scr (mL/min) (EQUATION 35.1):
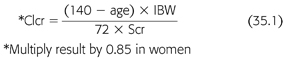
Patients with acute kidney injury should have an assumed Clcr of less than 10 mL/min, and it should be remembered that CG Clcr overestimates the GFR in these setting and should be avoided for assessment of kidney function in acute kidney injury. There are a number of new method to estimate renal function in chronic kidney disease: Modification of Diet in Renal Disease (MDRD) and Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI). These new markers of GFR are currently being utilized in both the research and clinical setting to more accurately measure renal function without exposing the patient to radiolabeled material. It is important to mention that these formulas approximate GFR rather than Clcr and, therefore, may be a more accurate estimate of renal function. It has been shown to be more accurate than other formulas for predicting GFR. The formula uses a creatinine assay (the kinetic alkaline picrate reaction), which is the least subject to artifact interference. GFR was predicted over a wide range of values including variables for ethnicity and serum albumin concentration and did not rely on timed urine collections. The MDRD study equation (16) is as follows (EQUATION 35.2):
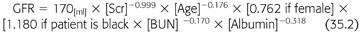
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


