 BIOCHEMICAL ABNORMALITIES IN PATIENTS WITH CHRONIC KIDNEY DISEASE-MINERAL AND BONE DISORDER
BIOCHEMICAL ABNORMALITIES IN PATIENTS WITH CHRONIC KIDNEY DISEASE-MINERAL AND BONE DISORDER
The first component of CKD-MBD is abnormal mineral metabolism including that of serum calcium, phosphate, PTH, vitamin D, and FGF-23. The pathophysiology of CKD-MBD comprises a number of feedback loops involving the kidney, bone, intestine, heart, and the vasculature, with the aim of maintaining the calcium and phosphate balance, often at the expense of abnormalities in other components of the system. The kidney plays a central role in regulating mineral metabolism, and as such, disturbances in mineral metabolism may occur early in the course of CKD because of the inability of the failing kidneys to maintain the levels of serum phosphate and calcium in the normal range. Bone disease develops as early as CKD stage 2 and becomes almost universal in patients with CKD stage 5 (9,10). Vascular calcifications also develop early, and its prevalence increases as the GFR declines such that approximately 80% of incident dialysis patients have evidence of coronary artery calcification (11–13). Given that these abnormalities are interrelated, and that they worsen with progressive loss of GFR, a clear understanding of the central role of the kidney in calcium and phosphate homeostasis will certainly help in understanding the pathophysiology of CKD-MBD.
In order to document the various abnormalities relevant to CKD-MBD, laboratory evaluation of serum phosphate, calcium, PTH, alkaline phosphatase, as well as selective imaging for soft-tissue calcification should be done. In this regard, several studies have shown that serum calcium and phosphate values do not become abnormal until the GFR falls below 40 mL/min/1.73 m2 and are relatively stable until GFR falls below 20 mL/min/1.73 m2 (14,15). However, 12% of patients with GFR >80 mL/min/1.73 m2 had a high PTH (defined as >65 pg/mL, the upper limit of normal of the assay used), and almost 60% of patients with GFR <60 mL/min/1.73 m2 had elevated PTH levels (14). In incident dialysis patients, serum levels of calcium and phosphate at the start of dialysis were 9.35 mg/dL and 5.23 mg/dL, respectively, but increase over the initial 6 months of renal replacement therapy (16). High levels of serum alkaline phosphatase greater than 120 U/L were reported in almost 30% of prevalent dialysis patients and found to be associated with mortality (17). In this chapter, these abnormalities are discussed, and current therapeutic options are summarized.
The Role of Phosphorus in Chronic Kidney Disease-Mineral and Bone Disorder
Phosphate is an inorganic molecule with a central phosphorus atom and four oxygen atoms. Although the terms phosphorus and phosphate are often used interchangeably, the term phosphate refers to the sum of the two physiologically occurring inorganic ions, hydrogen phosphate (HPO42−) and dihydrogen phosphate (H2PO4−), but most laboratories report this measurable, inorganic component as phosphorus. Inorganic phosphorus is critically important for a number of physiologic functions including cell membrane phospholipid content and function, cell signaling, energy transfer through mitochondrial metabolism, platelet aggregation, mineral metabolism, and skeletal development. As a result, the body has developed homeostatic mechanisms to maintain serum phosphate in the range of 2.5 to 4.5 mg/dL.
Phosphorus is the sixth most abundant element in the human body. The adult human body contains between 700 and 1,000 g of phosphorus, of which 85% is in bone, 14% is in the intracellular compartment, and the remaining 1% is in the extracellular fluids (FIGURE 24.2) (18). Normal daily dietary intake varies from 800 to 1,500 mg. Absorption of dietary phosphate occurs along the entire length of the small intestine, but absorption occurs mainly in the small intestine (19,20). Intestinal phosphate absorption occurs through two well-defined mechanisms (21). Most of the phosphate absorption occurs by a passive, concentration-dependent diffusion process. It is still unclear whether this passive absorption takes place by a paracellular or transcellular transport pathway. Of note, this pathway for sodium-independent phosphate transport is unregulated (22). Even in patients with severe hyperphosphatemia, phosphate absorption from the diet is only slightly lower than that in normal subjects. A smaller fraction of phosphorus is absorbed through a saturable, active transport pathway through the intestinal epithelial brush border sodium phosphate cotransporter 2b (Npt2b) which is regulated by dietary phosphate and 1,25 dihydroxyvitamin D [1,25(OH)2 D3] (21,22). These transporters are expressed in the small intestine and are upregulated under conditions of dietary phosphate deprivation. It is this active phosphate transport mechanism that becomes important under conditions of low phosphate intake. For example, during dietary phosphate restriction, 80% to 90% of phosphorus is absorbed. Approximately 150 mg of phosphorus is secreted daily into the gastrointestinal (GI) tract.
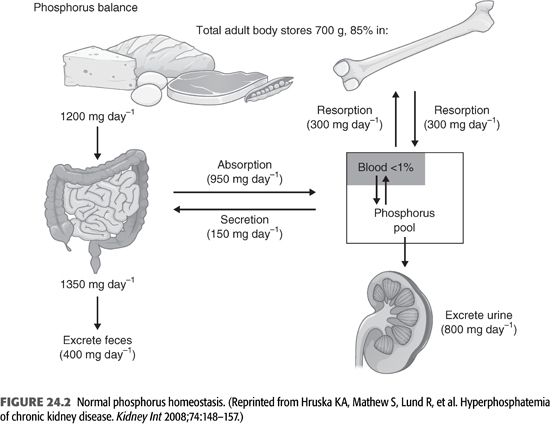
To maintain phosphate balance, the kidneys are responsible for excreting an amount of phosphorus equals to that absorbed from the intestine. The main factors that regulate proximal tubular phosphate excretion are the serum phosphate concentration itself, PTH, as well as FGF-23. These factors act on the proximal tubular epithelial brush border sodium phosphate cotransporters (Npt2a and Npt2c). Thus, serum phosphate concentration is primarily determined by the ability of the kidneys to excrete dietary phosphate. Interestingly, there is a diurnal variation in both serum phosphate levels and urinary phosphate excretion. Serum phosphate levels reach a nadir in the early hours of the morning, increasing to a plateau at 4 PM, and further increasing to a peak from 1 to 3 AM (23,24). A similar circadian pattern of serum phosphate was reported in CKD with lowest concentrations at 8 AM and highest at 4 PM and 4 AM (25). However, no diurnal variation was found in patients on dialysis when studied on a nondialysis day (26).
Phosphorus is present in most food products, particularly those with high protein content (27,28). In general, the higher the protein intake, the higher the phosphorus load, particularly in foods with a high phosphate-to-protein ratio as well as those with food additives and preservatives (27,20). In the steady state, serum phosphate concentration is primarily determined by the ability of the kidneys to excrete dietary phosphate (29). However, a tendency toward phosphate retention due to decreased phosphate filtration and excretion occurs as the GFR declines, but hyperphosphatemia is not commonly seen until the GFR drops to <25 to 40 mL/min/1.73 m2. This is due to the actions of PTH, FGF-23, and klotho on the proximal tubular phosphate reabsorption. In patients with advanced kidney disease, hyperphosphatemia results from a decrease in functioning nephrons, a decrease in the amount of filtered phosphate, and a subsequent decrease in excreted phosphate. Thus, in patients with CKD stage 4 or 5, the dietary intake of phosphate will likely exceed urinary excretion which leads to a positive phosphate balance. Hyperphosphatemia occurs almost universally in patients undergoing dialysis despite an apparent dietary phosphate restriction. Initially, excess phosphate will be buffered by bone, but once bone buffering is saturated, soft tissue phosphate deposition, usually in combination with calcium, ensues.
Consequences of Hyperphosphatemia
High serum phosphate is not a benign biochemical abnormality as it has been shown to be associated with a number of adverse clinical consequences (FIGURE 24.3). In this review, I will focus on the effects that hyperphosphatemia exerts in CKD-MBD. In the parathyroid gland (PTG), hyperphosphatemia directly stimulates parathyroid cells leading to increased PTH secretion. Also, it suppresses the 1-α-hydroxylase activity in the kidney which contributes to calcitriol deficiency. Finally, hyperphosphatemia stimulates the secretion of FGF-23 from osteocytes as will be discussed later.
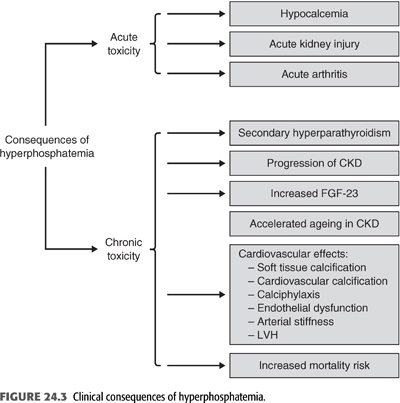
Perhaps, the most important consequence of hyperphosphatemia is on the cardiovascular system where it stimulates osteoblastic transformation of the vascular smooth muscle cell (VSMC) in the vasculature and directly contributes to extraskeletal calcification. This was supported by finding an association of high serum phosphate with increased mortality risk was demonstrated in patients with CKD, particularly those on maintenance dialysis. Epidemiologic studies in the last two decades have consistently identified serum phosphate as an independent predictor of cardiovascular mortality in dialysis patients (2,30,31). In 25,588 patients on hemodialysis from Dialysis Outcomes and Practice Patterns Study (DOPPS), serum phosphate level of 3.6 to 5 mg/dL was associated with the lowest risk of cardiovascular mortality, while serum phosphate level greater than 7 mg/dL was associated with the greatest risk of mortality (32). Serum phosphate levels are also associated with cardiovascular outcomes in non-CKD individuals. Higher serum phosphate, even within the normal range, was found to be associated with a greater risk of carotid atherosclerosis, coronary calcification, and cardiovascular mortality in patients with CKD (33,34). Moreover, in 15,732 patients from the Atherosclerosis Risk in Communities (ARIC) Study followed for 12.6 years, serum phosphate adjusted for GFR was associated with coronary events, stroke, and cardiovascular death (35). Higher serum phosphate is also associated with arterial stiffness, a marker of cardiovascular risk that may be related to vascular calcification (36). A recent meta-analysis of 47 cohort studies in 327,644 patients with CKD found an 18% increased risk of death for every 1-mg/dL increase in serum phosphate, while there was no significant association between all-cause mortality and serum PTH or calcium (6). Even in individuals with GFR >70 mL/min, higher serum phosphate levels were associated with a greater risk of cardiovascular events in a 15-year follow-up (37). However, these epidemiologic studies suffer from a number of limitations such as the level of serum phosphate above which there is increased risk of mortality in dialysis and CKD patients and the circadian variability in serum phosphate levels, which can be as high as 1 mg/dL, making interpretation of serum phosphate levels in the aforementioned studies difficult (24). Also, no clinical trial has shown that lowering serum phosphate to any specific level is associated with improvement in patients’ outcomes. Nonetheless, a number of pathophysiologic mechanisms underlie the association between higher serum phosphate and CVD. These include cardiovascular calcification, endothelial dysfunction, and left ventricular hypertrophy (LVH) (see below).
The Role of Phosphate in Secondary Hyperparathyroidism
It has been recognized for decades that persistent hyperphosphatemia plays a key role in the pathogenesis of secondary hyperparathyroidism (SHPT). The mechanism by which phosphate plays such a role are both indirect and direct. Elevations in serum PTH levels may become evident usually when the GFR falls below 60 mL/min/1.73 m2 before hyperphosphatemia or hypocalcemia are detectable by routine laboratory measurements. To explain this phenomenon, it was initially proposed that reduced GFR leads to retention of phosphate, transiently increasing serum phosphate level which in turn leads to a reduction in the level of serum ionized calcium in the blood, which then triggers an increase in the secretion of PTH. The goal for the increase in PTH level is to restore the serum calcium back to normal. Thus, PTH increases renal phosphate excretion by decreasing proximal tubular resorption of phosphate, mobilizing calcium from bone, and by stimulating the production of calcitriol by the kidney, which in turn increases intestinal absorption of calcium. As a result, a new steady state is reached where serum phosphate and calcium are normal at the expense of a higher level of PTH. This “trade-off” continues as the GFR declines further until severe SHPT develops (38). In this regard, several studies have shown that high-phosphate diet results in parathyroid hyperplasia (39,40). Moreover, dietary phosphate restriction in proportion to the reduction in GFR was effective in preventing the initial rise in PTH secretion and the development of hyperparathyroidism (41,42). Beside the reciprocal decline in serum ionized calcium due to transient hyperphosphatemia, phosphate retention may also inhibit the 1-α-hydroxylase enzyme in the kidney and thus may decrease the production of calcitriol. Moreover, it is now clear that high serum phosphate may directly stimulate the production and secretion of PTH, independent of its effects on serum ionized calcium and calcitriol (42–45). This effect is posttranscriptional and suggests that the stability of PTH mRNA may be regulated by phosphate and that this effect is mediated by proteins within the PTG that bind to the 3′ untranslated region of the PTH gene transcript (46–49). Furthermore, phosphate retention seems to reduce the production of arachidonic acid by parathyroid tissue; this signaling mechanism may be due to the effect of changes in cytosolic calcium on the phospholipase A2–arachidonic acid pathway (50). On the other hand, the effect of a low-phosphate diet in preventing parathyroid growth seems to be mediated by an increase in the cell cycle regulator p21 (51). Therefore, there are both indirect and direct mechanisms by which serum phosphate initially promotes PTH secretion (FIGURE 24.3). However, as will be discussed later, the recent discovery of FGF-23 has changed our understanding of the pathogenesis of SHPT.
The Role of Serum Calcium in Chronic Kidney Disease-Mineral Bone Disorder
Dietary calcium is also absorbed by two different mechanisms. First is an active, transcellular pathway via the transient receptor potential vanilloid 6 (TRPV6) channel on the apical membrane of the duodenum and proximal jejunum, and second by a paracellular pathway that occurs throughout the length of the intestine (52,53). Of the average 1,000-mg calcium ingested by a normal adult per day, 800 to 900 mg of these appear in the feces. Of the ingested calcium, 400 to 500 mg may be absorbed and 300 mg is secreted via the intestinal secretions, thus leaving a net calcium absorption of only 100 to 200 mg per day (54). In healthy adults who are in calcium balance, this net amount of absorbed calcium is excreted in the urine. The adult human body contains approximately 1,200 g of calcium, 99% of which is stored in bone as calcium–phosphate complexes, primarily as hydroxyapatite. Skeletal mineralization is one of the most important functions of calcium as it provides skeletal strength and a dynamic store to maintain the intra- and extracellular calcium pools. Less than 1% of the total body calcium is present in the serum, and its concentration is tightly regulated within a narrow physiologic range that is optimal for its functions. This range is between 8.5 and 10.5 mg/dL, but serum calcium is not a good indicator of the total body calcium. Moreover, it should be realized that the ionized component, which represents 40% of the total serum calcium, is the fraction of serum calcium that is closely regulated. In persons with healthy kidneys, normal serum level of calcium is maintained through the interaction of PTH and 1,25(OH)2 D3 (calcitriol), the active metabolite of vitamin D. Decreased ionized calcium level stimulates secretion of PTH which in turn acts to increase serum calcium (55).
The kidneys play a critical role in the regulation of normal serum calcium concentration which is maintained in the normal range until very late in CKD when it may decrease slightly. Although calcium balance in CKD is poorly understood, two recent balance studies in patients with CKD not yet on dialysis have shed some light on this subject. In the first study, Spiegel and Brady (56) found that subjects with late stage 3 and stage 4 CKD were in slightly negative to neutral calcium balance on a daily calcium intake of 800 mg but were in markedly positive balance on 2,000 mg calcium intake per day. In the second study, Hill et al. (57) studied eight patients with stage 3 or 4 CKD who received a controlled diet with or without 1,500 mg/d calcium carbonate supplement during two 3-week balance periods in a randomized placebo-controlled crossover design. The study found that patients were in neutral calcium balance while consuming a diet containing 957 mg calcium per day. However, increasing calcium intake with 500 mg calcium from calcium carbonate taken three times daily with meals produced positive calcium balance. The authors attributed this positive calcium balance to increased intestinal calcium absorption and net retention in the face of the kidneys’ failure to increase urine calcium excretion. As will be discussed later, the adverse consequences of positive calcium balance on mortality and cardiovascular calcification continue to be hotly debated.
Effects of Calcium on the Parathyroid Hormone
Since calcium plays a critical role in a wide range of biologic functions, its serum concentration is maintained within a very narrow range by a number of hormones including PTH, vitamin D, FGF-23, calcitonin, and estrogen. Among these, PTH is the primary regulator of serum calcium. On the other hand, ionized calcium is the primary regulator of PTH secretion (FIGURE 24.4). This bidirectional relationship has been described by an inverse sigmoidal curve (58). The midpoint or set point of this curve is the calcium-stimulated PTH, which is a key determinant of the serum ionized calcium concentration in vivo. In normal subjects, a small decrease in serum ionized calcium results in a sharp increase in serum PTH level within minutes. Also, an equally small increase in serum ionized calcium results in rapid decrease in the serum PTH level. Decreased serum ionized calcium triggers a sharp increase in PTH secretion which in turn acts to increase the plasma calcium by activating the 1-α-hydroxylase enzyme in the kidney and increasing the synthesis of calcitriol [1,25(OH)2 D3], which in turn enhances intestinal calcium absorption. PTH also mobilizes calcium from the readily available bone calcium stores and activates bone resorption indirectly by binding to PTH receptors on osteoblasts, which in turn increase osteoclast number and activity but also directly by binding and stimulating osteoclasts which lead to bone resorption (59). Finally, PTH increases calcium reabsorption in the distal nephron by activating adenylyl cyclase, which increases cyclic AMP levels and protein kinase A. The latter then phosphorylates and activates the transient receptor potential vanilloid 5 (TRPV5) which leads to increased transcellular calcium transport. Moreover, PTH increases the expression of several calcium transport proteins such as TRPV5, calbindin-D28k, and other transport proteins in the distal nephron, which also enhance renal calcium reabsorption (60). As a result, free serum calcium concentration is maintained in the normal range.
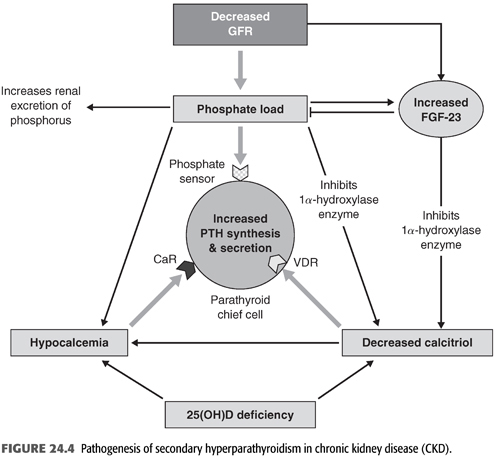
Total serum calcium usually decreases during the course of CKD. This is due to several factors including phosphate retention, decreased calcitriol [1,25(OH)2 D3] level, and resistance to the calcemic actions of PTH on bone. Phosphate retention may cause calcium precipitation in soft tissues, decreases calcium efflux from bone by increasing resistance of bone to PTH action, and inhibits the 1-α-hydroxylase enzyme, leading to further reduction in the already decreased level of calcitriol and decreased intestinal absorption of calcium (61). However, hypocalcemia is relatively uncommon in CKD patients and may not be critical in the development of SHPT (62). Moreover, the increase in PTH level first become evident when the GFR drops below 60 mL/min/1.73 m2, despite normal serum calcium and phosphate concentrations (15). On the other hand, circulating calcitriol concentrations begin to fall much earlier when the GFR is even above 60 mL/min/1.73 m2 and are markedly decreased in subjects with ESKD (63). The primary reason for the decline in calcitriol concentration is increased FGF-23 concentration, rather than the loss of functioning renal tissue (64). Hyperphosphatemia may also contribute to the decline in calcitriol synthesis late in the course of CKD by suppressing the 1-α-hydroxylase enzyme. Thus, the primary stimulus for PTH secretion in CKD is the low calcitriol level. However, when hypocalcemia is present and persistent, it directly increases PTH mRNA concentrations via posttranscriptional actions and stimulates the proliferation of parathyroid cells over days or weeks leading to the development of PTG hyperplasia (65).
Changes in serum ionized calcium are sensed by the calcium-sensing receptor (CaSR) on parathyroid cells. More than two decades ago, it was shown that the secretion of PTH is mainly regulated by the CaSR, a membrane receptor that responds to small changes in serum ionized calcium (66). This receptor, a member of the G-protein-coupled membrane receptors on the parathyroid chief cells, enables the parathyroid cells to act as calcium electrodes and thus sense and respond rapidly to very small changes in the level of extracellular calcium. Hypocalcemia stimulates PTH secretion by inactivating the CaSR while hypercalcemia activates the CaSR and inhibits the secretion of PTH. Also, CaSR inhibits PTH gene expression at the transcriptional and/or posttranscriptional level, and thus, it can inhibit parathyroid cellular proliferation. The discovery and cloning of the CaSR in the PTG has increased our awareness of the importance of calcium ion in regulating PTH secretion. Kovacs et al. (67) have shown that CaSR knockout mice develop severe parathyroid hyperplasia and hyperparathyroidism despite significantly increased 1,25(OH)2 D3 levels. By contrast, studies in vitamin D receptor (VDR) knockout mice show that these VDR-deficient animals develop hyperparathyroidism that can be reversed by the administration of calcium and normalizing serum calcium levels (68). Therefore, it was not surprising that CaSR became a rational target for therapeutic intervention by agents that either mimic the actions of calcium ion on PTH secretion (type I calcimimetics) or induce changes in the structural conformation of the CaSR and thus increase the sensitivity of the receptor to extracellular ionized calcium (type II calcimimetics) and can lower PTH secretion from the PTG. In CKD, the number of CaSRs may be reduced in hypertrophied PTGs, particularly in areas of nodular hypertrophy (69–71). Decreased expression of the CaSR appears to be related to the proliferation of parathyroid tissue, and both may be related to increased serum phosphate (70,72). Such decrease in CaSR number can lead to inadequate suppression of PTH secretion by calcium, resulting in inappropriately high PTH concentrations in the setting of normal or even high calcium concentrations. However, experimental studies suggest that calcimimetic agents may even suppress PTH secretion in human parathyroid cells with pathologically reduced CaSR levels (73) and attenuate parathyroid hyperplasia (74).
Role of Vitamin D in Chronic Kidney Disease-Mineral Bone Disorder
Calcitriol [1,25(OH)2 D3] is a key hormone in the regulation of mineral metabolism. It acts by binding to a nuclear VDR, which is ubiquitously distributed in most tissues, leading to transcriptional regulation of target genes. Calcitriol is a critical factor in the regulation of serum calcium, phosphate, PTH, and FGF-23. It enhances the absorption of phosphate from the small intestinal cells by upregulating the apical membrane sodium-dependent phosphate cotransporter 2b (Npt2b), the rate-limiting step in intestinal phosphate absorption. It also helps to maintain the plasma calcium concentration through actions on the intestine, bone, and the kidney. In the intestine, it acts by increasing the expression of TRPV6 while in the kidney, it enhances calcium reabsorption by increasing the expression of the transporter protein TRPV5 in the distal renal tubular and connecting tubular cells (75). However, its effect on bone with regard to regulating serum calcium is not entirely clear but may involve increasing calcium release from bone by binding to osteoblasts and osteocytes and suppressing bone matrix mineralization during negative calcium balance (76).
CKD is both a state of calcitriol deficiency and resistance. Nutritional vitamin D deficiency (25-hydroxyvitamin D), which is very common in CKD, is an important contributor to calcitriol deficiency (64,77). In general, 25-hydroxyvitamin D is not biologically active, and thus, it must be metabolized in the kidney to its active form, the 1,25(OH)2 D3 or calcitriol, by the enzyme 1-α-hydroxylase in the renal proximal tubular cells and several other extrarenal cells. There are a number of factors that regulate the activity of this enzyme, the most important of which is PTH, serum phosphate, serum calcium, and various hormones (FIGURE 24.4). More recently, FGF-23 and secreted frizzled related protein 4 (sFRP4) have been shown to inhibit the 1-α-hydroxylase enzyme activity (78,79). In fact, it has been shown that the primary cause of the low calcitriol level in patients with CKD is increased FGF-23 concentration rather than the loss of functioning renal tissue (64). Also, hyperphosphatemia may contribute to the decline in calcitriol synthesis late in the course of CKD by suppressing the 1-α-hydroxylase enzyme. Given that calcitriol exerts a potent negative feedback on the PTG, thereby decreasing PTH production and release, its low level in CKD is now believed to trigger the initial increase in PTH secretion. However, when hypocalcemia is present and persistent, it directly increases PTH mRNA concentrations via posttranscriptional actions and stimulates the proliferation of parathyroid cells over days or weeks leading to the development of PTG hyperplasia (65).
Role of Parathyroid Hormone
PTH (1-84) is a single-chain polypeptide of 84 amino acids that is cleaved from pre-pro-PTH, where it is stored along with its fragments in secretary granules in the PTG. PTH is synthesized as a pre-pro-PTH and then is proteolytically processed to pro-PTH in the endoplasmic reticulum and then to PTH in the Golgi and secretory vesicles. All intracellular pro-PTH is normally converted to PTH before secretion. Once released, PTH has a short half-life of 2 to 4 minutes as it is cleaved after secretion into N-terminal, carboxy C-terminal, and midregion fragments. These fragments are usually metabolized by the liver and in the kidney (80). In patients with reduced GFR, the renal excretion of the C-fragment is reduced, resulting in marked elevation of PTH levels. PTH 1-84 is the biologically active hormone that exerts its effects through the interaction of its first 34 amino acids with the type 1 parathyroid hormone receptor (PTHR1). Some of PTH fragments lacking residues at the extreme N-terminus such as PTH (7-84) is believed to interact with its own distinct receptors and may have antagonistic effects to the calcemic action of PTH (1-84) and PTH (1-34) (81,82).
The serum concentrations of PTH depend on its release from stores in secretory granules within the PTG and also by synthesis of new PTH. The primary stimulus for PTH secretion is reduction in serum ionized calcium concentration. Hypocalcemia, acting through the CaSR, induces a rapid release of stored PTH in secretory granules and retards its degradation within the PTG. Moreover, it increases division of the parathyroid cells in the PTG likely through the action of the CaSR (55). In addition to calcium, high serum phosphate levels also increase PTH secretion independently of changes in serum calcium or serum levels 1,25(OH)2 D3. However, it is not clear how the parathyroid cell senses changes in the serum phosphate level. Also, vitamin D alters the transcription of PTH and may indirectly inhibit its release by increasing the expression of CaSR in the PTG (FIGURE 24.4). Low vitamin D level removes the inhibitory effect of vitamin D on transcription and thus stimulates PTH synthesis and secretion. On the other hand, hypercalcemia, administration of the active vitamin D analogs, as well as high FGF-23 level inhibit PTH synthesis or release (55,83,84).
PTH is considered a systemic toxin that has been implicated in the pathogenesis of various uremic complications including CKD-MBD (85). It acts by binding to its receptor, the PTHR1, which is widely expressed in many tissues including bone, kidney, and the vasculature. As discussed above, PTH increases extracellular fluid calcium by mobilizing calcium from its readily available stores in bone and also by increasing osteoclastic bone resorption. Moreover, it increases calcium reabsorption in the distal nephron by upregulating the TRPV5 and TRPV6, calbindin-D28K, and other transport proteins in the distal nephron (60). Another important function of PTH is its phosphaturic effect which is mediated by rapid internalization and subsequent degradation of Npt2a and Npt2c proteins in the apical membrane of the proximal tubule. PTH also stimulates the 1-α-hydroxylase enzyme in the kidney which in turn converts 25OHD into its active form, the 1,25(OH)2 D3. Lastly, PTH enhances the synthesis and secretion of FGF-23 from bone cells. Studies have shown that continuous infusion of (1-34) PTH for 24 to 46 hours increased serum FGF-23 levels in healthy volunteers and in patients with ESKD (86,87). By contrast, parathyroidectomy decreased serum FGF-23 levels (88,89). However, since PTH stimulates the production of 1,25(OH)2 D3, it is difficult to determine whether its effect on FGF-23 is the result of a direct stimulatory effect on osteocytes or to an indirect effect of elevated 1,25(OH)2 D3 levels (90). Finally, because of its effect on bone, both high and low PTH levels may increase the risk of vascular calcification (91). The systemic toxic effects of PTH are many and include cardiomyopathy, hyperlipidemia, carbohydrate intolerance, bone marrow fibrosis, and anemia resistant to erythropoietin, pruritus, peripheral neuropathy, and encephalopathy (85,92,93). Moreover, high PTH serum levels have been shown to be associated with higher mortality rates in dialysis patients (2,31,94). In a recent study from DOPPS, the mortality risk in dialysis patients was higher for PTH level of 301 to 450 pg/mL and >600 pg/mL compared with the group with PTH of 150 to 300 pg/mL. Also, cardiovascular mortality and the risk of hospitalization were higher for PTH >600 pg/mL. On the other hand, PTH level <50 pg/mL was also associated with mortality, particularly among patients with diabetes and those with lower body mass index (95).
Accurate measurement of the serum PTH concentration is essential for the proper evaluation and management of patients with CKD-MBD. Measurements and interpretation of PTH values in patients with CKD, particularly those on chronic dialysis, remain challenging with wide variations among various assays in current use (96). What constitutes an appropriate assay for PTH measurement remains unclear. PTH level was initially measured by radioimmunoassay (RIA) (first-generation assays) using various polyclonal antibodies directed against epitopes that are located within the mid- or C-terminal portion of the PTH molecule. However, these assays proved to be inaccurate and have been replaced by two-site immunometric assay (IMAs) (second- and third-generation assays). The second-generation assays, so-called “intact PTH” assays, not only measure the full-length PTH molecule (1-84) but also detect the PTH (7-84) fragment (97,98). On the other hand, the more recent third-generation assay measures only PTH (1-84) and is referred to as the whole, bioactive or biointact PTH assay. However, the role of this assay in the diagnosis of renal osteodystrophy remains to be established.
The difficulty in interpreting PTH values led some authors to suggest that measurements of PTH levels in CKD and dialysis patients are not even necessary because there is inadequate evidence to link PTH to skeletal and cardiovascular end points in CKD (80). However, others believe that there is sufficient evidence that links PTH with adverse outcomes, especially when levels are toward the extremes of the KDIGO recommendations, and thus, PTH measurements are helpful in evaluating CKD-MBD in such patients (99). In our opinion, a single value of PTH measured at a single time point is unlikely to precisely reflect the underlying bone disease in patients with CKD, but the trend of serial measurements of PTH, using the same PTH assay, would be more informative. Another important but difficult issue is what represents an appropriate target range for PTH level. The 2009 KDIGO clinical practice guidelines suggest a broad range of PTH values, ranging from 2 to 9 times the upper limit of normal (7). Although very high or very low PTH values can predict with some certainty the nature of the underlying bone disease, we believe that the higher end of the KDIGO recommended value for PTH is bound to increase the prevalence of osteitis fibrosa (100). Indeed, a recent study from DOPPS showed that SHPT is on the rise since the KDIGO guidelines were released (95). Moreover, data from the Control of Renal Osteodystrophy in South America study and the Analyzing Data, Recognizing Excellence, and Optimizing Outcomes study from Europe showed that PTH values as recommended by the 2003 KDOQI guidelines may be associated with the best clinical outcomes (101,102). Thus, as we previously suggested, a PTH level in the 100 to 400 range may minimize both low turnover bone disease and osteitis fibrosa (100). This range is also supported by recent data from the Japanese Society for Dialysis Therapy (JSDT) which showed that patients with PTH levels outside their recommend target range of intact PTH between 60 and 240 pg/mL have increased mortality risk (100,103). The adverse consequences of high PTH levels was recently reported from the Cardiovascular Health Study which showed that serum PTH levels >65 pg/mL were associated with greater N-terminal pro-B-type natriuretic peptide, cardiac troponin T, and left ventricular mass, particularly in patients with CKD (104).
Role of Fibroblast Growth Factor-23 in Chronic Kidney Disease-Mineral Bone Disorder
FGF-23 is a circulating peptide that plays a key role in the control of serum phosphate concentrations (105,106). FGF-23 is produced by bone osteocytes and osteoblasts in response to calcitriol, increased dietary phosphate load, PTH, and calcium (106,107). The primary function of FGF-23 is to maintain normal serum phosphate concentration by reducing renal phosphate reabsorption and by reducing intestinal phosphate absorption through decreased calcitriol production (FIGURE 24.4). In the renal proximal tubular cells, FGF-23 binds to the fibroblast growth factor receptor (FGFR) and its coreceptor, klotho, causing inhibition of the expression of the Npt2a and possibly Npt2c (108). As discussed earlier, FGF-23 also inhibits expression of 1-α-hydroxylase enzyme, leading to decreased calcitriol synthesis by the kidney (109).
FGF-23 and PTH mutually regulate each other in a negative feedback loop (110). Studies have shown that PTH stimulates synthesis and secretion of FGF-23 by activation of PTH/PTHrP receptor on osteocytes and osteoblasts (88,90,111). On the other hand, other studies showed that FGF-23 suppresses PTH synthesis (112,113). However, among CKD patients, the presence of high PTH concentrations, despite high FGF-23 concentrations, suggests that the PTG is relatively resistant to the elevated concentrations of FGF-23 in uremia. This may be related to the markedly decreased expression of FGFR 1 and klotho protein in the hyperplastic PTG (114,115). Klotho is a transmembrane protein that is produced by osteocytes and is essential for FGF-23 receptor activation (116). Klotho is expressed in the kidney and the PTGs, making these organs the target for FGF-23. Unlike FGFRs which are quite ubiquitously distributed, klotho expression is restricted, and thus, its presence may control whether a cell is an FGF-23 target. Its expression in the kidney and PTGs decreases during CKD progression in mice and humans (113,114,117). Klotho itself is a target gene of calcitriol (118,119). Moreover, klotho extracellular domain does not directly bind to FGF-23 but enhances FGF-23 binding to its receptor complex with a much higher affinity than to the FGFR alone (120). Decreases in klotho induce resistance to FGF-23 in these target organs, which further increases blood FGF-23 levels (113,114,117). It has been shown that a 50% decrease in klotho expression results in approximately 300% increase in blood FGF-23 levels in mice (121).
Patients with CKD have increased FGF-23 concentrations due to increased secretion by osteocytes and decreased catabolism by the diseased kidney (122,123). Klotho expression declines early in the course of CKD and then progressively with decreasing GFR (124). The reduction in klotho temporally coincides with the rise in FGF-23, suggesting that this decline may be partially responsible for the progressive rise in FGF-23 concentration. Moreover, the decrease in klotho expression on hyperplastic PTGs may contribute to the resistance and impaired parathyroid suppression by FGF-23 (114). On the other hand, FGF-23 may be a regulator of klotho expression in the kidney (121). Klotho deficiency is bound to decrease its regulation of FGF-23 production and thus allows hyperphosphatemia to become the principal regulator of FGF-23 secretion in CKD. Moreover, the decrease in membrane-bound klotho expression curbs FGF-23 actions on FGFR/klotho complexes leading to loss of negative feedback to FGF-23 secretion and continued production of FGF-23 by the osteocyte. Interestingly, in the absence of klotho expression as in the heart, the very high levels of FGF-23 in advanced stages of CKD enable FGF receptor activation in the heart, independent of klotho, leading to direct pathologic effects such as left ventricular hypertrophy (125).
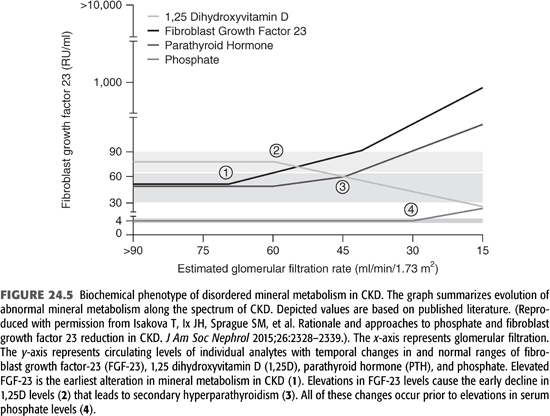
FGF-23 level increases prior to changes in calcium, phosphorus, calcitriol, or PTH levels and therefore is now recognized as one of the earliest detectable biomarkers of the CKD-MBD (FIGURE 24.5) (126). Patients with advanced CKD have increased FGF-23 and PTH levels but decreased calcitriol levels. This constellation of compensatory changes initially result in normal serum calcium and phosphate levels, but eventually, such mechanisms become overwhelmed in patients with ESKD resulting in the full expression of the syndrome of CKD-MBD. One component of CKD-MBD that FGF-23 and/or klotho may play a role is vascular calcification. Some studies reported an independent association between circulating FGF-23 level with the progression of coronary calcifications (127,128), but more recent data indicate that FGF-23 is not involved in vascular calcification (129), while klotho deficiency is a recognized factor in that process (129). However, robust data are currently available to implicate the high levels of FGF-23 in dialysis and CKD patients with left ventricular mass, heart failure, and mortality but not with ischemic heart disease (125,130–132).
 CARDIOVASCULAR CALCIFICATION
CARDIOVASCULAR CALCIFICATION
The second important component of CKD-MBD is vascular calcification. This type of calcification results from abnormal deposition of calcium phosphate salts into the vascular wall as well as in other cardiac structures including the aorta, coronary arteries, cardiac valves, as well as peripheral vessels. Vessels from healthy control subjects do not calcify even after long-term exposure to supraphysiologic levels of phosphate and/or calcium in vitro, whereas vessels from CKD and dialysis patients calcify (133). Calcification starts as early as CKD stage 2, as shown in an animal model of CKD equivalent to human CKD stage 2 (134) but becomes accelerated with progressive loss of GFR particularly in patients with ESKD who are receiving dialysis. Several studies have documented that patients with ESKD on dialysis have a higher prevalence and severity of cardiovascular calcification than age- and gender-matched healthy subjects. Vascular calcification in patients with CKD can be either atherosclerotic affecting the intima, or medial calcification with calcium phosphate deposition in the media of the vessel wall. It has been argued that medial calcification is a manifestation of accelerated atherosclerosis in patients with CKD (135), but others believe that atherosclerotic and medial calcification are two distinct entities, with the latter developing more commonly in patients with CKD particularly those with diabetes mellitus (136). Clearly, both forms of calcification can occur concomitantly in patients with CKD.
The cause of accelerated calcification in patients with ESKD is multifactorial and include both the traditional CVD risk factors and uremia-related risk factors (12,137). Vascular calcification in patients with CKD was initially thought to be a passive, physicochemical process in which serum phosphate and calcium are deposited in the arterial wall. However, we now know that vascular calcification is more complex and involves an active, cell-mediated, and highly regulated process similar to bone formation with VSMC playing a critical role. Just as in bone formation, vascular calcification results from either an imbalance between factors that promote calcification and those regulatory molecules that act to inhibit calcification (12,137,138). Incubation of human aortic smooth muscle (SM) cells in high phosphate medium similar to those regularly seen in dialysis patients directly enhanced extracellular calcification in these cells via a sodium-dependent phosphate cotransporter-sensitive mechanism (139). In this scenario, serum phosphate induces transformation of VSMC to an osteoblast-like cell by simultaneously downregulating the production of SM genes such as α-actin and SM22 and upregulating Runx2 (Cbfa1), osterix, osteopontin, osteocalcin, and alkaline phosphatase. Similarly, high calcium in the medium also induces transformation of VSMC to an osteoblast-like cell (133). It is likely that calcium and phosphate have synergistic procalcification effects in mediating VSMC cell apoptosis, osteochondrocytic differentiation, and matrix vesicle release (140). However, recent in vitro studies have found calcium to be a stronger procalcification factor than phosphate. Experimental studies utilizing human VSMC explant cultures or aortic ring cultures clearly document a role for calcium in the pathogenesis of vascular calcification in CKD and dialysis patients. In one study, when arterial rings obtained during abdominal surgery from children with normal renal function, those with impaired renal function, and from those on dialysis were exposed to high phosphate and calcium media, calcium-induced calcification more potently than phosphate, but only in rings from patients with CKD or those on dialysis (133). This suggests that VSMCs from healthy vessels have effective inhibitory mechanisms that prevent calcification, while those with CKD, particularly dialysis patients, become susceptible to calcification.
The now phenotypically transformed VSMCs lose their contractile function and behave like bone-forming cells that lay down a collagen-1 rich extracellular matrix and pinch-off matrix vesicles that are rich in calcium and phosphate. These vesicles are capable of initiating mineralization of the vascular wall just as they do so in bone (138). The key transcription factor here is Runx2 without which no such transformation of VSMCs or mineralization of the vessel wall can occur. Increased calcium levels promote calcification further by inducing apoptosis of VSMCs (133,141,142). Both matrix vesicles and apoptotic bodies serve as nucleation sites for hydroxyapatite, particularly in the presence of low calcification inhibitors such as matrix-Gla protein (MGP) and fetuin-A. Thus, unlike vessels from healthy individuals, vessels from predialysis and, to a much greater extent, dialysis patients have been primed to calcify (133). Susceptibility to calcification in CKD may be related to a decrease in matrix vesicles calcification inhibitors such as fetuin-A and MGP and to degradation of the elastin by matrix metalloproteases (MMP), such as MMP-2 and MMP-9, which are upregulated in the arterial wall in CKD (138). The degradation of elastin leads to overexpression of transforming growth factor (TGF)-β, which is thought to be involved in osteoblast differentiation and in enhancing VSMC calcification and arterial stiffness (138). From the above, it can be deduced that vascular calcification is the result of interaction between factors that promote calcification and those that normally inhibit calcification in the microenvironment of the vessel wall and not simply by spontaneous deposition of calcium phosphate.
One of the factors that was suspected of enhancing vascular calcification in dialysis and CKD patients is the calcium load provided from use of calcium-based phosphate binders (CBPBs). While a number of clinical trials (143–145), epidemiologic studies (146), and meta-analysis (147) have shown increased risk of all-cause mortality or progression of cardiovascular calcification in CKD patients treated with CBPB, other clinical trials (148,149), epidemiologic studies (150), and meta-analysis (151) did not show a difference between calcium-based and non–calcium-based phosphate binders. In fact, a recent randomized placebo-controlled pilot clinical trial by Block et al. (152) showed that therapy with both non–calcium- and calcium-based phosphate binders resulted in progression of vascular calcification, albeit to a smaller degree in noncalcium phosphate binders. Thus, the effects of calcium load from use of CBPB on progression of cardiovascular calcification, while plausible, remain controversial (153). An important issue that needs to be investigated is whether slowing vascular calcification by any means, including phosphate binders, would translate into improvements in clinical outcomes. Two epidemiologic studies have in fact reported that use of any type of phosphate binder, possibly with the exception of aluminum-based binders, is independently associated with improved survival in dialysis patients compared with no use of binder at all (154,155). A third study in an incident United States Renal Data System (USRDS) cohort that started dialysis in 1996 to 1997, when only CBPBs were used in the United States, found no association between the use of these agents and mortality (156). As a result, the KDIGO Work Group stated that “there were inconclusive data to indicate that any one binder has beneficial effects on mortality or other patient-centered outcomes when compared with any other binder” (7). However, 16 of 17 members of the KDIGO Work Group felt that the weight of evidence is in favor of the non-CBPBs. In patients with CKD stages 3 to 5D, the Work Group recommend restricting the dose of CBPBs and/or the dose of calcitriol or vitamin D analog in the presence of persistent or recurrent hypercalcemia, arterial calcification, or adynamic bone disease (ABD).
The clinical consequences of vascular calcification in patients with CKD are serious. Cardiovascular calcification increases the risk of cardiovascular events including myocardial infarction, fatal arrhythmia, congestive heart failure, and valvular heart disease (4,157). Hyperphosphatemia may be associated with increased cardiac stroke index, increased systolic and decreased diastolic blood pressure, increased pulse pressure, and increased pulse wave velocity (4,157). Moreover, these patients had higher common carotid artery diameter and lower wall-to-lumen ratios. The authors speculated that these changes were due to medial calcification of the arterial wall, which leads to arterial stiffness. These hemodynamic disturbances may result in LVH and compromise coronary artery blood flow during diastole. It is noteworthy that in the Framingham Heart Study, abdominal aortic calcification identified by plain radiographs was independently predictive of subsequent vascular morbidity and mortality (158).
 RENAL OSTEODYSTROPHY
RENAL OSTEODYSTROPHY
The third component of CKD-MBD is renal bone disease. Abnormal bone lesions are common in patients with CKD and usually start early when most biochemical measures of mineral metabolism are still normal. Most patients with early stages of CKD (stages 1 to 3) have age-related osteoporosis, while those with the more advanced stages of CKD (stages 3 to 5D), particularly those who have abnormalities of mineral metabolism, usually have renal osteodystrophy (159). Abnormal bone quality and quantity can lead to increased bone fragility, resulting in fracture. Both osteoporosis and renal osteodystrophy may coexist and can lead to increased bone fragility and fractures in all stages of CKD.
In patients with CKD, progressive bone lesions may result in bone pain, fractures, and deformities in growing children, reduced growth velocity, and abnormal height, particularly in those on maintenance dialysis. The prevalence of fractures among the dialysis population is between 10% and 40% (7). Such complication is often associated with serious consequences. As an example, hip fractures may be associated with bleeding, infection, and increased risk of mortality. Osteoporosis is traditionally diagnosed by finding low bone mineral density (BMD). By contrast, CKD-MBD can lead to an abnormal bone quality even in the setting of a normal or high bone mineral content in patients with CKD stages 3 to 5D. Thus, measurement of BMD by dual-energy x-ray absorptiometry (DXA) scan may not distinguish between the various types of renal osteodystrophy as seen with bone histology (7). A more accurate but also more expensive is the use of quantitative computed tomography (QCT), which was recently shown to prospectively identify more bone loss at the hip than DXA (160). However, bone biopsy is the gold standard as it provides information on the three variables suggested by KDIGO: bone turnover, mineralization, and volume (TMV). Turnover refers to a spectrum of bone formation rates from abnormally low to very high. Mineralization reflects the amount of unmineralized osteoid. Bone volume, which is the end result of changes in bone formation and resorption rates, contributes to bone fragility. In a recent review of studies dating from 1969 to 2007, the KDIGO group reported that bone volume/trabecular volume (BV/TV) is generally lower in dialysis patients compared with that in nondialysis CKD patients across all renal osteodystrophy categories (7).
Renal osteodystrophy encompasses four different types of bone lesions including osteitis fibrosa cystica, ABD, osteomalacia, and mixed lesions (TABLE 24.1). The prevalence of these types of bone diseases, as determined by bone biopsy carried out in adult dialysis patients between 1983 and 2006, showed that osteitis fibrosa was present in 34% of patients, ABD in 19%, osteomalacia in 10%, and mixed lesions in 32%. Only 2% of bone biopsies in dialysis patients were reported as normal (7). However, the prevalence of these bone lesions have changed over the last two to three decades, largely due to changes in patients’ demographics and in therapeutic strategies. In the early dialysis era, aluminum-based phosphate binders were the predominant phosphate binders, vitamin D therapy was not routinely used, dialysis patients were younger, diabetes was not as prevalent, and multiple renal transplantations were not frequently done. Also, clinical studies of the relationship between plasma PTH levels and bone histology among dialysis patients during the 1980s and early 1990s reflected the impact of the changes in therapeutic strategies and the demographics of the dialysis population during that period. Of note, nearly all studies during this period used a first-generation RIA for PTH from Nichols Institute Diagnostics. In the present era, more than half of patients receiving dialysis are 65 years of age or older, and nearly 50% of them have diabetes, and one-third of diabetic patients are African Americans. Moreover, aluminum-based phosphate binders were replaced by CBPB and more recently with non–calcium-containing binders such as sevelamer hydrochloride and lanthanum carbonate. Also, large intermittent intravenous doses of vitamin D analogs as well as cinacalcet are being used to treat the bone lesion associated with SHPT. Consequently, aluminum-induced bone disease has decreased from 40% of biopsies carried out before 1995 to 20% in patients biopsied after 1995 (7).

Unfortunately, few studies have assessed the impact of these demographic and therapeutic changes on bone histology among patients with CKD. However, in a seminal paper, Sherrard et al. (161) reported the results of their bone biopsy study in 259 patients from their dialysis program. Their results showed an increased incidence of the so-called aplastic bone lesion, particularly in patients treated with peritoneal dialysis. Sixty-six percent of these patients had low turnover bone lesions, whereas 62% of those treated with hemodialysis had the high turnover bone lesion of SHPT. Aluminum-associated osteomalacia was noted less frequently than before, largely due to the substantial decline in the use of aluminum-based phosphate binders. This spectrum of bone lesions is considerably different from that previously reported. Indeed, in hemodialysis patients, the frequency of osteitis fibrosa and ABD are currently similar, but ABD is the predominant lesion in peritoneal dialysis patients. Yet, the increasing use of non–calcium-containing phosphate binders and cinacalcet may result in a change in the histologic bone lesions in the future. Moreover, the higher target for PTH level recommended by the KDIGO clinical practice guidelines may also result in resurgence of predominant lesions of osteitis fibrosa (7).
 PATHOPHYSIOLOGY OF RENAL BONE DISEASE
PATHOPHYSIOLOGY OF RENAL BONE DISEASE
High Bone Turnover Bone Disease
Osteitis Fibrosa and Secondary Hyperparathyroidism
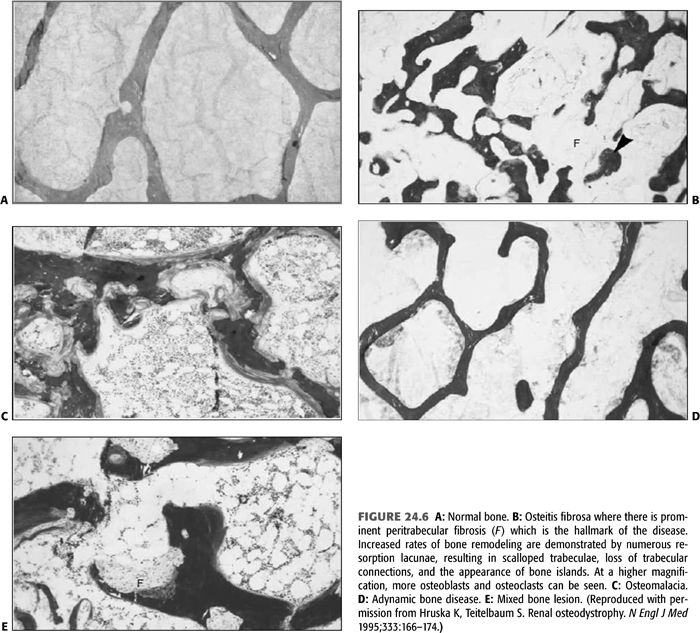
Bone is the main reservoir for both calcium and phosphate, and its turnover is regulated by PTH and vitamin D. Thus, disturbances in these mineral metabolism are expected to result in bone disease. Normal bone histology is illustrated in FIGURE 24.6A. The classical histopathologic form of renal bone disease is osteitis fibrosa, an entity caused mainly by SHPT (TABLE 24.1 and FIGURE 24.6B). The disease begins quite early in the course of CKD when the GFR declines to less than 60 mL/min/1.73 m2, in response to a series of abnormalities that initiate and maintain increased PTH secretion (162,163). The hallmark of osteitis fibrosa is marrow fibrosis and increased bone remodeling (164). There is increased bone formation rate, which is characterized by increased amount of osteoid, and increase in the number of osteoblasts. Concomitantly, there is increased resorption of bone caused by increased number and activity of osteoclasts. Many of the histologic features occur in response to the high PTH level with contributions from local cytokines and low level of calcitriol. The degree to which these features develop is generally proportional to the PTH level and the duration of exposure to such levels. In the aforementioned study, Sherrard et al. (161) noted that 45 of their 259 unselected patients had mild and 57 had severe hyperparathyroidism. Those with more severe disease had a longer duration of renal failure. As discussed above, a number of factors play a role in triggering increased synthesis and secretion of PTH by the PTGs in patients with CKD. These include decreased serum ionized calcium, phosphate retention, decreased plasma levels of 1,25(OH)2 D3 (calcitriol), the active form of vitamin D, and skeletal resistance to the calcemic actions of PTH.
The Role of Ionized Calcium in Secondary Hyperparathyroidism
PTH level undergoes diurnal variations, primarily due to changes in serum ionized calcium which modulate the minute-to-minute secretion of PTH. Consequently, ionized calcium concentration remains maintained in the normal range in CKD, largely due to mobilization of calcium from bone by PTH (165). Total serum calcium usually decreases during the course of CKD. This is due to several factors including phosphate retention, decreased 1,25(OH)2 D3 level, and resistance to the calcemic actions of PTH on bone. Hypocalcemia is relatively uncommon in CKD patients and may not be critical in the development of SHPT (62). However, when present and persistent, it enhances the pre-pro-PTH gene transcription and ultimately promotes the development of PTG hyperplasia. Phosphate retention may cause calcium precipitation in soft tissues, decreases calcium efflux from bone by increasing resistance of bone to PTH action, and inhibits the 1-α-hydroxylase enzyme, leading to further reduction in the already decreased level of calcitriol and decreased intestinal absorption of calcium (61).
More than two decades ago, it was shown that the secretion of PTH is mainly regulated by CaSR, a membrane receptor that responds to small variations in serum ionized calcium (66). The CaSR is a member of the G-protein-coupled membrane receptors that is highly expressed in multiple tissues including the surface of the chief cells of the PTG, kidneys, bone marrow, osteoclasts, osteoblasts, and others (166). It enables the parathyroid cells to sense very small changes in the level of extracellular ionized calcium and rapidly responds to bring about the required changes in PTH secretion. In turn, by increasing bone resorption, stimulating the formation of the active vitamin D (calcitriol) in the kidney and by reducing renal calcium excretion, PTH acts to raise the serum ionized calcium concentration toward normal. In the kidney, CaSR, which is expressed on the basolateral membrane on the cells of the thick ascending limb of the loop of Henle, is a major regulator of urinary calcium excretion (167,168). In addition to calcium, the CaSR can also be activated by magnesium and certain amino acids (169). In summary, CaSR orchestrates changes in the PTG and kidney functions in order to normalize serum calcium concentration. Consequently, CaSR became a target for therapeutic intervention for SHPT and osteitis fibrosa. Calcimimetic agents are now routinely used in clinical practice for treatment of SHPT in patients with ESKD (170).
The Role of Phosphate Retention in and Secondary Hyperparathyroidism
As explained earlier, hyperphosphatemia does not become evident until stage 4 CKD, a tendency toward phosphate retention due to decreased phosphate filtration and excretion occurs as the GFR declines (15,171). Elevations in serum PTH levels may become evident when the GFR falls below 60 mL/min/1.73 m2 before hyperphosphatemia and hypocalcemia are detectable by routine laboratory measurements. Initially, the “trade-off” hypothesis was proposed to explain this phenomenon, where progressive decline in renal function leads to a new steady state where serum phosphate and calcium are normal at the expense of a higher level of PTH. This “trade-off” continues as the GFR declines further until severe SHPT develops. In this regard, several studies have shown that high phosphate diet results in parathyroid hyperplasia (39,40). Moreover, dietary phosphate restriction in proportion to the reduction in GFR was effective in preventing the initial rise in PTH secretion and the development of hyperparathyroidism (41,42). The mechanism by which phosphate retention plays a role in the pathogenesis of SHPT includes both indirect and direct actions. Beside the reciprocal decline in serum ionized calcium due to transient hyperphosphatemia, phosphate retention may also inhibit the 1-α-hydroxylase enzyme in the kidney and thus may decrease the production of calcitriol. Moreover, it is now clear that phosphorus may directly inhibit the production and secretion of PTH, independent of its effects on serum ionized calcium and calcitriol (43–45). This effect is posttranscriptional and suggests that the stability of PTH mRNA may be regulated by phosphorus and that this effect is mediated by proteins within the PTG that bind to the 3′ untranslated region of the PTH gene transcript (46,47,49). Furthermore, phosphate retention seems to reduce the production of arachidonic acid by parathyroid tissue; this signaling mechanism may be due to the effect of changes in cytosolic calcium on the phospholipase A2–arachidonic acid pathway (50). On the other hand, the effect of a low-phosphorus diet to prevent parathyroid growth seems to be mediated by an increase in the cell cycle regulator p21 (51).
The Role of Calcitriol
Decreased production of calcitriol in CKD patients removes the inhibitory action of calcitriol on the PTG and leads to increased production of PTH. In turn, high PTH level enhances the activity of 1-α-hydroxylase enzyme in the kidney and tends to increase the plasma level of calcitriol back to the normal range. This action is counter balanced by several other factors: First and most important is the increased level of the phosphaturic hormone FGF-23 in patients with CKD which decreases the production of calcitriol by suppressing the activity of 1-α-hydroxylase enzyme (64,172). Second, phosphate retention resulting from decreased renal function contribute to suppression of 1-α-hydroxylase enzyme and decreased calcitriol production. Third, decreased level of 25(OH)D, the precursor for calcitriol is very common in CKD patients (64,173). Finally, decreased delivery of 25(OH)D that is bound to the vitamin D–binding protein into the cell. This compound is filtered through the glomerulus and is reabsorbed in the proximal tubules by the endocytic receptor megalin. Endocytosis is required to preserve 25(OH)D and to deliver it into the cell for generation of 1,25(OH)2 D3. In CKD, there is decreased delivery of 25OHD to the 1-α-hydroxylase enzyme, particularly in presence of proteinuria due to loss of vitamin D–binding protein in the urine (174). These events lead to decreased intestinal absorption of calcium and hypocalcemia, which in turn stimulates PTH secretion. However, in vitro and in vivo studies have shown that calcitriol directly inhibits PTH synthesis by acting through the VDR in parathyroid cell nuclei leading to decreased pre-pro-PTH messenger RNA in a dose-dependent manner (175,176). In contrast, calcitriol deficiency may lead to increased PTH production, even in the absence of overt hypocalcemia, by removing its direct suppressive action on PTH secretion. A decrease in calcitriol concentrations also lowers the number of VDRs in the parathyroid cells in experimental animals and in uremic patients (177–180). Moreover, the role of uremic toxins in inhibiting calcitriol receptor binding to vitamin D response elements may contribute to the calcitriol resistance of renal failure (181,182).
The Role of Parathyroid Gland Structure and Function
One of the earliest changes in the PTG during the course of CKD is parathyroid hyperplasia. This structural change develops within few days following experimental CKD and increases in parallel with the progression of renal disease (183). Factors that are involved in triggering hyperplasia are still unclear but include persistent hypocalcemia, calcitriol deficiency, and hyperphosphatemia (184,185). Hypocalcemia is a potent stimulus for PTG growth, and its effect is mediated by the CaSR (184). Moreover, studies have suggested that the set point for calcium-regulated PTH secretion is abnormal in the hyperplastic PTG (186). This is likely due to decreased expression of the CaSR in the hyperplastic PTG of CKD patients (69,187). The CaSRs themselves may contribute to PTG hyperplasia because the use of calcimimetic agents have been shown to prevent PTG hyperplasia (74,170,188).
Decreased calcitriol levels may also contribute to parathyroid hyperplasia. Pertinent to this mechanism is that VDR expression is downregulated in hyperplastic PTG (178,180) and administration of calcitriol is associated with upregulation of expression of VDRs as well as the CaSRs (176,189). The severity of parathyroid hyperplasia in patients with CKD correlates directly with the reduction of PTG VDR (180). Prolonged PTG stimulation initially leads to diffuse polyclonal hyperplasia, but in patients in whom hyperplasia was allowed to continue, monoclonal expansion of the parathyroid cells develops, leading to the formation of nodules within the PTG (190–192). Studies in genetically modified mice indicate that signal transduction via the CaSR is a key determinant of parathyroid cell proliferation and PTG hyperplasia (193). It seems that parathyroid cell hyperplasia precedes the downregulation of CaSR expression in a uremic rat model (194). When nodular hyperplasia is present, there is decreased expression of both VDRs and CaSRs. This in turn leads to increased set point for the concentration of calcium that is needed to decrease PTH secretion and the response of the PTG to therapeutic maneuvers becomes more difficult leading to refractory SHPT.
The Role of Skeletal Resistance to the Actions of Parathyroid Hormone
Patients with CKD are known to have reduced calcemic response to PTH and thus require a higher PTH level to maintain normal bone turnover (195,196). Thus, in order to keep a normal bone turnover in patients with CKD, the level of PTH must be 2 to 3 times its normal level. On the other hand, excessive suppression of PTH secretion will lower the bone turnover (197). This skeletal resistance to the calcemic action of PTH was demonstrated in patients with acute kidney injury, CKD patients acutely infused with PTH, early CKD patients with high PTH level, advanced CKD, hemodialysis patients, and renal transplant recipients with decreased renal function. The pathogenesis of skeletal resistance to PTH is multifactorial and includes phosphate retention (198), downregulation of the PTH receptors (199,200), low levels of calcitriol (201–203), and inhibition of PTH binding to receptors by the 7-84 PTH fragment (81). Other factors include increased level of osteoprotegerin (OPG), which acts as a decoy receptor for osteoclastogenesis-promoting factor in patients with CKD (204,205) and decreased level of BMP-7 (206). The calcemic response to high PTH may be restored after adequate amounts of both 1,25(OH)2 D3 and 24,25(OH)2 D3 (207).
Low Bone Turnover Bone Disease
Adynamic Bone Disease and Osteomalacia
The low turnover lesions are categorized into osteomalacia and ABD. Osteomalacia is characterized by very low rate of bone turnover and marked accumulation of unmineralized osteoid (FIGURE 24.6C). Until a decade ago, the most common cause of osteomalacia in patients with ESKD was aluminum toxicity which is known to inhibit bone mineralization. However, the incidence of osteomalacia has decreased with the abandonment of aluminum-based phosphate binders and the introduction of more efficient techniques for treatment of water used in preparing the dialysate. Another possible cause of osteomalacia in CKD patients is deficiency of 25-hydroxyvitamin D.
On the other hand, ABD is being seen with increasing frequency in dialysis and CKD patients, particularly those with diabetes (161,208–210). Moreover, variations in its prevalence can be seen in different geographic regions (211,212). ABD is characterized by low bone turnover in the absence of aluminum overload. The KDIGO Work Group examined bone biopsy studies between 1983 and 2006 and found the prevalence of ABD to be 18% in CKD stages 3 to 5 not yet on dialysis, 19% in hemodialysis patients, and 50% in peritoneal dialysis patients (7). In these studies, ABD has replaced osteomalacia and exceeded that of osteitis fibrosa (213). Even in patients with advanced CKD not yet on dialysis, the prevalence of low turnover disease has reportedly increased to between 12% and 23% (10,214). Definitive diagnosis of ABD requires bone biopsy showing an overall reduction in cellular activity in bone, including both the number of osteoblasts and osteoclasts (FIGURE 24.6D), which leads to very low rate of bone turnover. In contrast to osteomalacia, ABD is characterized by low bone turnover without osteoid accumulation. Moreover, both the rate of collagen synthesis by osteoblasts and its subsequent mineralization are subnormal (208,213).
The pathogenesis of ABD is poorly understood but likely involves multiple factors (215,216). In some patients, bone lesions may be irreversible as seen in patients with diabetes mellitus and those associated with osteoporosis due to old age, menopause, or steroid therapy. ABD may also be caused by the malnutrition–inflammation–cachexia syndrome (MICS) (217). However, in other patients, the bone lesions are reversible and are amenable to therapeutic maneuvers. This is particularly true in patients who develop the disease due to aluminum toxicity; parathyroidectomy; and treatment with large doses of CBPBs, calcitriol, or the use of high dialysate calcium, factors suspected of oversuppressing PTH secretion. High doses of calcitriol may also have PTH-independent inhibitory effects on osteoblastic proliferation and function and thus may induce ABD even in presence of high PTH levels. Interestingly, oxacalcitriol has been shown to control SHPT in uremic dogs without inducing low bone turnover (218). Patients undergoing peritoneal dialysis are particularly at increased risk of developing ABD due to sustained exposure to high calcium concentration in the dialysate. Use of low dialysate calcium concentration or non–calcium-containing binders have been reported to lead to reversal of ABD lesion (219,220). An additional pathogenetic mechanism for ABD was also described. Studies in animal models showed that CKD directly produces ABD by diminishing skeletal growth factors or by producing anabolic inhibitors or both (221). Interestingly, ABD developed when CKD was induced despite the presence of SHPT. Pertinent to this mechanism is the fact that ABD has been described in CKD patients before dialysis (222,223). The mechanism by which CKD causes ABD is not clear but may involve several factors such as uremic toxins, acidosis, increased concentration of OPG and N-terminally truncated PTH fragments, prior use of steroid therapy, malnutrition, and disturbances in growth factors and cytokines. The importance of growth factors in the pathogenesis of ABD was demonstrated in experimental animals in that treatment with the anabolic factor bone morphogenetic protein (BMP)-7 reversed the adynamic bone lesions (224).
Most patients with ABD are asymptomatic, although some may develop bone pain (208,216). Moreover, patients with ABD are at increased risk of fractures and hypercalcemia (225–227). Importantly, patients with ABD are at increased risk of vascular calcification. One study in hemodialysis patients (228) reported an association between low bone turnover and vascular calcification. Another study by the same authors found a significant interaction between the dose of CBPBs and bone activity such that calcium load had a significantly greater impact on aortic calcification and stiffness in patients with ABD (229).
Other Bone Lesions
Mixed bone lesions are now commonly encountered in bone biopsies of patients with CKD. This was observed in 18 of the 259 patients reported by Sherrard et al. (161). The lesion is characterized by an increase in unmineralized osteoid as seen in osteomalacia and an increase in marrow fibrosis similar to that found in the severe hyperparathyroidism (FIGURE 24.6E). Bone formation is increased in this group as a result of elevated PTH level. Sherrard et al. (161) speculated that the high PTH level in patients with this bone lesion was almost certainly due to inadequate calcium replacement as lack of adequate calcium may result in a large amount of unmineralized osteoid. Therefore, under these conditions, although calcium may have been deposited at a very high rate, the overstimulated osteoblasts were forming osteoid at an even higher rate giving rise to the mixed bone lesion. This entity has also been described with a normal or low bone formation. Such a picture may be seen when aluminum toxicity and hyperparathyroidism coexist and produce normal bone formation or even a low bone formation if aluminum toxicity dominates the picture.
Dialysis-related amyloidosis (DRA) is another bone lesion that results from amyloid deposits in articular and periarticular tissues in dialysis patients, particularly in those who have been receiving dialysis for over 10 years (230,231). This entity is not caused by abnormalities in mineral metabolism and has no effect on bone morphology or vascular calcification. As a result, it is not currently included in the KDIGO clinical practice guidelines for the diagnosis, evaluation, prevention, and treatment of CKD-MBD. DRA is a disabling disease characterized by accumulation and tissue deposition of amyloid fibrils consisting of β2-microglobulin in the bone, periarticular structures, and viscera of patients with CKD (208). This is because reduced renal clearance of this protein in patients with decreased GFR leads to plasma accumulation and slow tissue deposition. β2-Microglobulin amyloid deposits may also be found at subperiosteal locations (232). There are a number of risk factors associated with DRA including older age, dialysis vintage, use of low-flux dialysis membranes and/or bioincompatible dialysis membrane, and lack of residual renal function (233,234).
Patients with DRA usually complain of shoulder pain related to scapulohumeral periarthritis and rotator cuff infiltration by amyloid. Also, patients may develop symptoms of carpal tunnel syndrome due to amyloid deposition in the carpal tunnel which causes hand pain and numbness and dysesthesias in the distribution of the median nerve. Carpal tunnel syndrome is a recognized complication of amyloid deposits and develops frequently in patients on long-term hemodialysis therapy (235). Large cystic deposits can be seen in the long bones, clavicles, phalanges, carpal or tarsal bones, and pelvis. Fractures through these cysts or into infiltrates in the femoral head are particularly common and heal with difficulty. Of particular note, these large, often cystic deposits are commonly misdiagnosed by radiologists as the “brown tumors” of osteitis fibrosa cystica. In fact, cysts, particularly large cysts, are distinctly uncommon in SHPT. When hyperparathyroid cysts do occur, they are more likely to be found in the mandible or skull, and are usually quite small (230,232). Renal transplant is the most effective treatment modality for amyloid-induced symptoms, but the use of biocompatible dialysis membranes may also be helpful (236). Beside enhancing β2-microglobulin clearance by dialysis or renal transplantation, treatment of DRA is otherwise palliative. Analgesics help with periarticular and bone pain, but since DRA is a progressive disease, early surgical correction of CTS is warranted.
Diagnosis of Renal Osteodystrophy
Clinical Presentation
Renal bone disease is usually asymptomatic, particularly in the earlier stages of CKD. Even in advanced stages of CKD, symptoms are often nonspecific and include bone pain, arthralgia, proximal muscle weakness, and spontaneous tendon rupture (TABLE 24.2). These symptoms are not helpful in differentiating between SHPT and low turnover bone diseases. However, patients with low turnover disorders, not due to aluminum, have increased incidence of asymptomatic hypercalcemia. Moreover, in one study, 34% of the patients with ABD but without significant aluminum deposition who were reevaluated after 5 years developed bone pain and fractures and had increased mortality (209).

Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


