Series
Time period
Age range
Total number of tumors
Gastrointestinal tumors
Finnish Cancer Registry (Teppo et al. 1975)
1953–1970
0–15
2,605
33 intestinal cancers, site not specified, 15 of these were lymphomas
Hacettepe University Children’s Hospital, Ankara, Turkey (Buyukpamukcu et al. 1996b)
1972–1994
0–17
6,774
5 carcinoids
4 “APUDomas”
3 leiomyosarcomas
16 colorectal carcinomas
2 gastroduodenal carcinomas
Northern Region Young Person’s Malignant Disease Registry (Northern England) (Cotterill et al. 2000)
1968–1995
0–25
4,316
28 colorectal cancers
7 small bowel cancers (lymphomas not broken down by site)
Columbus Children’s Hospital (Columbus, Ohio) (Bethel et al. 1997)
1952–1996
0–17
4,547
41 lymphomas
9 carcinoids
3 colon cancers
2 GIST
The Children’s Hospital of Philadelphia (tumor registry-unpublished data)
1965–2002
0–21
8,149
14 lymphomas
8 carcinoids
4 adenocarcinomas
In a recent analysis of data collected by the Surveillance, Epidemiology, and End Result (SEER) Program of the National Institutes of Health covering the years 1973–2005, a total of 375 patients aged 20 or younger were identified as having tumors (both benign and malignant) of the upper and lower gastrointestinal tracts. The overall incidence was 0.131 cases per million. In the upper gastrointestinal tract, 105 tumors were identified. 8.6 % of tumors occurred in the esophagus, 61 % in the stomach, and 30.5 % in the small bowel (Zhuge et al. 2009). In the colon and rectum a total of 270 cases were identified, and of these the most common histologies were adenocarcinoma (35.6 %) and carcinoid tumors (34.1 %) (Yang et al. 2010).
As scarce as the epidemiologic data is concerning pediatric gastrointestinal carcinoma, there is even a greater void when one attempts to examine the incidence of gastrointestinal mesenchymal tumors and polyps. The general experience of pediatric pathologists is that the vast majority of polyps and indeed the majority of tumors of the pediatric gastrointestinal tract are benign lesions. However, in children, more so than in adults, these lesions may be the sequelae or harbingers of more serious genetic, familial or developmental disorders, some of which may be premalignant. Recognition and management at an early age can be critical in the overall treatment of these patients and their families.
8.2 Polyps and Polyposis Syndromes
8.2.1 General Concepts
Intestinal polyps are grossly visible tumors which protrude into the lumen of the GI tract. They may be neoplastic, inflammatory, hyperplastic, or hamartomatous, and in children, the vast majority are benign. Apart from ruling out the rare case of malignancy within an individual polyp, it is of great importance to realize that these polyps may present in the context of a polyposis syndrome with predisposition to colorectal cancer or other malignancies. Salient features of polyposis syndromes are summarized in Table 8.2.
Table 8.2
Polyposis syndromes
Syndrome | Gene defect | Usual age when polyps manifest clinically | Histology | Polyp location | Risk of GI cancer |
|---|---|---|---|---|---|
Juvenile polyposis syndromes | SMAD4 BMPR1A | ||||
Juvenile polyposis coli | Childhood | Juvenile | Colon, occasional small bowel | Increased | |
Familial juvenile polyposis of the stomach | Childhood | Hyperplastic gastric | Stomach | Low | |
Juvenile gastrointestinal polyposis | Childhood | Juvenile | Colon, stomach, small bowel | Increased | |
Juvenile polyposis of infancy | Infancy | Juvenile | Colon, stomach, small bowel | Increased | |
Peutz-Jeghers syndrome | LKB1 (STK11) | Adolescence | Hamartomatous | Small bowel, colon, stomach | Increased |
Cronkhite-Canada syndrome | Unknown, probably not genetic | Adulthood | Juvenile (colon) Hyperplastic (stomach) | Stomach, small bowel, colon, esophagus | Low |
PTEN hamartoma syndrome | |||||
Cowden syndrome | PTEN | Adulthood | Mixed; juvenile and hamartomatous | Entire GI tract | Low |
Bannayan-Riley-Ruvalcaba syndrome | Adulthood | Mixed; juvenile and hamartomatous | Ileum, colon | Low | |
Familial adenomatous polyposis (including Gardner’s and Turcot’s syndrome) | APC | Adolescence | Adenomatous (colon) Hamartomatous (stomach) | Small bowel, stomach | Extremely high (colon) Increased in small bowel, stomach |
8.2.2 Juvenile Polyps and Juvenile Polyposis
8.2.2.1 Etiology
Juvenile or retention polyps account for about 90 % of polyps in children, by far the most common polypoid gastrointestinal lesion encountered. Although in some older reports, the terms juvenile polyp, inflammatory polyp, and hyperplastic polyp have been used interchangeably, they are actually separate entities, with different histologic and clinical characteristics. There is however considerable pathologic overlap between juvenile polyps and hamartomatous polyps, especially in their syndromic presentations. It is estimated that 1–2 % of children have one or more juvenile polyps, usually encountered between ages two and ten, with a peak incidence between ages two and five (Corredor et al. 2001). They are rarely encountered in infants under 1 year of age. In older reports, juvenile polyps have been described as usually solitary, with a predilection for the left colon. However, it now appears that a substantial number are multiple, and proximal colonic involvement is not uncommon. One study demonstrated 37 % of juvenile polyps to be proximal to the splenic flexure with 12 % of patients having only proximal colonic polyps (Gupta et al. 2001). In another study, 18 % of children undergoing colonoscopy for juvenile polyps had five or more (Corredor et al. 2001). It is now estimated that between 23 and 50 % of children with juvenile polyps have more than one polyp (Corredor et al. 2001; Gupta et al. 2001). This apparent change in the numbers and distribution of juvenile polyps may be a reflection of the increased use of pancolonoscopy in children.
When multiple juvenile polyps are present in an individual, juvenile polyposis syndrome (JPS) must be considered. Juvenile polyposis, which occurs at an incidence of approximately 1:100,000 individuals, is likely when 3–5 or more juvenile polyps are found or if any number of juvenile polyps (even one) are found in a patient with a family history of juvenile polyposis (Gammon et al. 2009). In most cases of juvenile polyposis, 50–200 polyps may be found; however, the number of polyps can be in the thousands, as illustrated in Fig. 8.1. In addition, the presence of extracolonic juvenile polyps is usually associated with a polyposis syndrome.
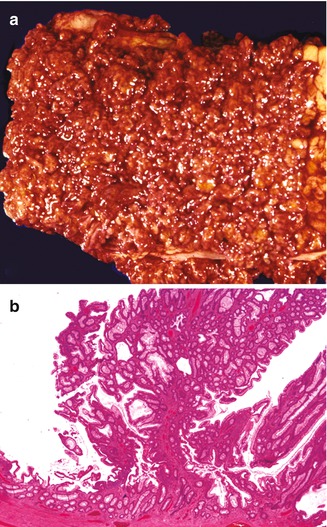
Fig. 8.1
Juvenile polyposis. (a) Portion of colon from an adolescent patient with juvenile polyposis, remarkable for the presence of innumerable polyps carpeting the mucosal surface. (b) Microscopic section of a polyp from the same specimen, demonstrating both cystically dilated glands and an arborizing architecture
There are three major clinical forms of juvenile polyposis: juvenile polyposis coli, juvenile gastrointestinal polyposis, and familial juvenile polyposis of the stomach. Juvenile polyposis coli is the most common of the three and is usually diagnosed in the first decade of life in children with multiple colonic polyps and occasionally small bowel juvenile polyps, but without polyps elsewhere in the alimentary tract. In juvenile gastrointestinal polyposis, polyps are found in the stomach and small bowel, in addition to the colon, and in familial juvenile polyposis of the stomach, juvenile polyps are limited to the stomach, where they consist of hyperplastic gastric glands and resemble gastric hyperplastic polyps. All these forms of juvenile polyposis can be familial. A fourth type, juvenile polyposis of infancy, is a rare juvenile polyposis that typically presents before age two and is often associated with death in the first year of life.
8.2.2.2 Genetics
The pathophysiology underlying the development of isolated juvenile polyps is not well established. No unique genetic abnormalities have been discovered, and solitary juvenile polyps appear to be a noninherited condition that is hamartomatous in nature (Giardiello and Offerhaus 1995). The tremendous inflammation seen in many of them suggests a contribution from inflammatory mediators. In one study, 12 of 28 juvenile polyps had foci of p53 reactivity, suggesting a potential for neoplastic transformation in addition to features of a reactive inflammatory process (Coffin and Dehner 1996).
Except for the rare juvenile polyposis of infancy, most cases of juvenile polyposis are suspected to be autosomal dominant, and approximately 50 % of patients have a positive family history. Approximately 50–60 % of cases of juvenile polyposis syndrome have been associated with germ-line mutations or deletions of either SMAD4 or BMPR1A, both of which are members of the TGF-β signal transduction pathway, which is integral to colonic cell cycle regulation (van Hattem et al. 2008; Woodford-Richens et al. 2000; Roth et al. 1999; Howe et al. 2001). In an interesting genotype-phenotype correlation, patients with SMAD4 gene abnormalities are more likely to have gastric polyps than patients with BMPR1A defects (Friedl et al. 2002). SMAD4 status of a juvenile polyp can be determined by immunohistochemistry, and a germ-line SMAD4 mutation is likely if SMAD4 expression is reduced or absent (Langeveld et al. 2010). PTEN mutations and deletions have also been described in juvenile polyposis; however, these cases may actually be forms of Cowden and Ruvalcaba syndromes (Corredor et al. 2001; Woodford-Richens et al. 2000; van Hattem et al. 2008). In addition, mutations of ENG, a gene associated with hereditary hemorrhagic telangiectasia, have been identified in several patients with juvenile polyposis (Sweet et al. 2005). However, as this finding has not been replicated, the role of ENG mutations in juvenile polyposis syndrome is still not established (Howe et al. 2007; van Hattem et al. 2008). Some individuals with juvenile polyposis also have hereditary hemorrhagic telangiectasia (HHT), with symptoms of telangiectasia, epistaxis, and anemia (secondary to gastrointestinal arteriovenous malformations). This combined form of juvenile polyposis and HHT appears to be associated with SMAD4 mutations (Gallione et al. 2004, 2006).
8.2.2.3 Clinical Manifestations
Juvenile polyps most commonly present with mild hematochezia, although bleeding can be profuse. The degree of blood loss can be sufficient to cause iron deficiency anemia, which is present in up to 1/3 of children with juvenile polyps (Corredor et al. 2001). Polyps in the distal colon can present clinically with tenesmus, diarrhea, and even rectal prolapse. It is not unusual for these polyps to autoamputate and be passed in the stool. When this occurs, the pathologist may be the end recipient of a markedly autolytic specimen collected at home by worried parents.
Individuals with juvenile polyposis tend to become symptomatic in childhood, as opposed to familial adenomatous polyposis, which is usually asymptomatic before adolescence (Grotsky et al. 1982). Rectal bleeding is the most common symptom, present in 84–95 % of patients, and can be life-threatening (Pashankar et al. 2000). Other symptoms may include diarrhea, protein-losing enteropathy, failure to thrive, and malnutrition. In juvenile polyposis of infancy, infants may present with severe disease heralded within the first month of life by bloody or mucoid diarrhea, and intussusception and rectal prolapse are not uncommon.
Juvenile polyposis may be associated with other alimentary tract and extraintestinal anomalies, such as hypertelorism, malrotation, hydrocephalus, undescended testes, and Meckel’s diverticulum (Table 8.3). Desai et al. detected extracolonic abnormalities in 78 % of their patients, including cardiac abnormalities and mental retardation (Desai et al. 1998). Some of these extracolonic manifestations may reflect underlying hamartomatous syndromes such as Cowden syndrome and Ruvalcaba syndrome (Corredor et al. 2001). Other associations include ganglioneuromatous proliferations in the gastrointestinal tract and hereditary hemorrhagic telangiectasia (see above) (Inoue et al. 1999). In addition to juvenile polyposis, juvenile polyps are seen in Cowden syndrome, Ruvalcaba syndrome, Cronkhite-Canada syndrome, and Gorlin syndrome (Woodford-Richens et al. 2000).
Table 8.3
Congenital abnormalities associated with juvenile polyposis
Digital clubbing |
Porphyria |
Macrocephaly/hydrocephalus |
Mental retardation |
Congenital heart disease |
Cleft lip/palate |
Malrotation of intestines |
Bifid uterus/vagina |
Undescended testes |
Hernias |
Hypertelorism |
Meckel’s diverticulum |
Supernumerary digits |
Pulmonary arteriovenous malformation |
Umbilical fistula |
Mesenteric lymphangioma |
Amyotonia congenita |
Despite isolated cases of colon carcinoma developing in solitary juvenile polyps (Jones et al. 1987), patients with solitary juvenile polyps do not have an increased risk of developing colon cancer compared to the general population (Kapetanakis et al. 1996; Giardiello et al. 1991). The earliest descriptions of juvenile polyposis held that this was not a premalignant condition, a view that has changed over the last several decades with a series of reports of adenomatous changes (Goodman et al. 1979; Mills and Fechner 1982; Jarvinen and Franssila 1984; Baptist and Sabatini 1985; Atsumi et al. 1991; O’Riordan et al. 1996) and the development of frank carcinoma (Longo et al. 1990; Jass et al. 1988), and it is now accepted that patients with juvenile polyposis have an increased risk of colorectal cancer (Coburn et al. 1995), as well as cancer of the small bowel. As opposed to solitary juvenile polyps, in which dysplasia is uncommon, dysplastic changes or coexisting adenomatous polyps are found in 8–31 % of patients with juvenile polyposis (Bentley et al. 1989; Wu et al. 1997). Patients with gastric polyps are at a higher risk of developing gastric carcinoma, particularly in patients with SMAD4 mutations (Gammon et al. 2009). The mean age of developing carcinoma in one series was 34–35 years (Jass et al. 1988).
8.2.2.4 Endoscopic Features
Isolated juvenile polyps typically have a smooth lobulated surface. In juvenile polyposis, the colon is usually peppered by 50 to hundreds of polyps, most of which are small, measuring between less than 1–5 mm in size. Most are pedunculated and not sessile. The intervening mucosa is generally unremarkable. Polyps from different parts of the gastrointestinal tract are similar in endoscopic appearance (Coffin and Dehner 1996).
8.2.2.5 Pathology
Most juvenile polyps have straightforward pathologic features and usually do not present a diagnostic problem. They may be lobulated and tend to have a red-tan, granular surface, which often is ulcerated. On sectioning, the polyps present an edematous glistening surface and often have macroscopic mucus-filled cysts (Fig. 8.2a).
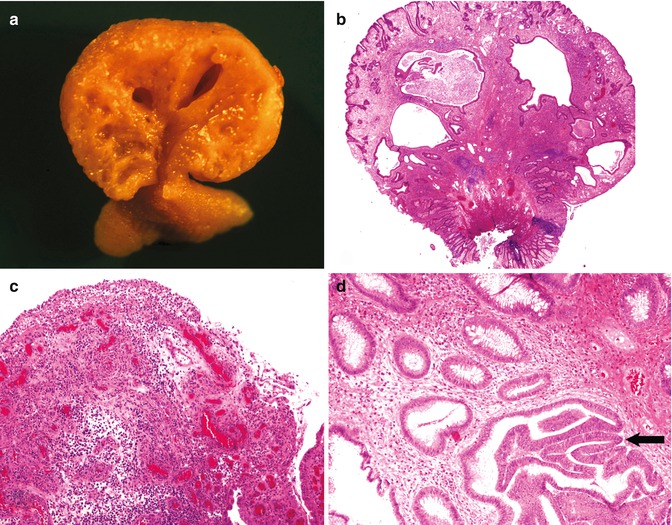
Fig. 8.2
Juvenile polyps. (a) Cut surface of a juvenile polyp. (b) Low-power view of a juvenile polyp illustrating cystically dilated glands and a chronically inflamed stroma. (c) Portion of a juvenile polyp histologically indistinguishable from a pyogenic granuloma. (d) Juvenile polyp with focal mild dysplasia (arrow)
Histologic examination discloses an edematous and prominent stroma in which variable numbers of cystically dilated glands are scattered (Fig. 8.2b). The glands may be branched or tortuous and often contain mucus. The background stroma is usually markedly inflamed, with a mixed inflammatory cell population including lymphocytes, plasma cells, eosinophils, and polymorphs. The epithelial surface may be cuboidal, flattened, or ulcerated. In some instances, the ulceration and inflammation is so extensive as to give an appearance of a pyogenic granuloma (Fig. 8.2c). The lamina propria is variably fibrotic and often contains ectatic prominent vessels and a stalk of smooth muscle; however, the smooth muscle component and degree of arborization is less than that seen in hamartomatous Peutz-Jeghers polyps, and juvenile polyps tend to have more architectural disorder. A serrated glandular pattern, similar to that seen in hyperplastic polyps, may be present.
Juvenile polyps may have evidence of regenerative atypia and even focal mild dysplasia, although significant dysplasia is rare in solitary juvenile polyps (Fig. 8.2d). Interpretive problems may arise when glandular epithelial cells within a polyp develop pseudostratification, crowding, nuclear hyperchromasia, and increased mitotic activity. These histologic findings are more common in larger polyps and have been referred to as epithelial atypia, dysplasia, and adenomatous change, but there is no consensus as to their significance (Coffin and Pappin 1997). Occasional findings include prominent hemorrhage, prominent lymphoid follicles, foreign body granulomas, metaplastic bone, and metaplastic cartilage (Groisman et al. 1994). Foci of ganglioneuromatous change have also been described (Pham and Villanueva 1989).
Individual colonic juvenile polyps in these juvenile polyposis conditions may differ pathologically from classic solitary juvenile polyps, with some having more arborization and a higher frequency of adenomatous change. In addition, recently published data suggests a correlation between germ-line gene defects and histology in juvenile polyps, with SMAD4 defects corresponding to higher polyp crypt density and BMPR1A defects corresponding to a more classic juvenile polyp morphology (van Hattem et al. 2008). Adenomatous changes may be seen in non-polypoid mucosa, as well as within the juvenile polyps themselves.
8.2.2.6 Treatment
Although there are no strict guidelines for the diagnosis and management of solitary juvenile polyps, removal of the polyps appears indicated, due to the high incidence of anemia and the possibility of adenomatous change. Further surveillance is generally thought unnecessary, unless symptoms recur or if the polyps have mixed adenomatous/juvenile histology.
Because of the heightened risk of developing colorectal neoplasia, it has been suggested that patients with three or more juvenile polyps or patients with a family history and any number of juvenile polyps should undergo regular surveillance for colorectal cancer (Giardiello et al. 1991). Management recommendations for juvenile polyposis include colonoscopy and upper endoscopy starting at age 15, repeated yearly if polyps are found and repeated every 2–3 years if the previous examination was normal. In cases where the number of polyps in the stomach or bowel is too great for endoscopic removal, surgical resection should be strongly considered (Zbuk and Eng 2007). Patients with SMAD4 mutations should also be screened for the presence of arteriovenous malformations, with subsequent periodic imaging.
Given the rarity of juvenile polyposis of infancy, no standard guidelines have been published.
8.2.3 The Peutz-Jeghers Syndrome
8.2.3.1 Etiology
The Peutz–Jeghers syndrome is an autosomally inherited syndrome of abnormal perioral pigmentation, multiple gastrointestinal polyps, and an increased incidence of benign and malignant neoplasms. First described in a set of twins in the late nineteenth century, the familial occurrence of the syndrome was first described by Peutz in 1921, and the classic description of the disease appeared in a landmark report by Jeghers in 1949 (McGarrity et al. 2000; Jeghers et al. 1949; Peutz 1921). The incidence of the syndrome is estimated to be 1 in 120,000 (McGarrity et al. 2000). Although some early investigators considered the polyps to be adenomatous, they are hamartomatous in nature, composed of multiple cell types native to their portion of the gut but aberrantly placed (McGarrity et al. 2000). Hamartomatous polyps develop in the stomach, small bowel, and colon, and the syndrome often remains undiagnosed until after these polyps are identified (Shepherd et al. 1987; Buck et al. 1992). Most frequently involved is the jejunum, followed by the ileum, duodenum, colon, and stomach, with sparing of the esophagus. Wide intrafamilial variability in clinical manifestations and cancer risk exist (McGarrity et al. 2000). By WHO criteria, in patients with a positive family history, the diagnosis is established by the presence of even one histologically confirmed Peutz-Jeghers polyp or by the presence of the characteristic mucocutaneous pigmentation. In the absence of a family history, the diagnosis can be established by finding three or more histologically confirmed Peutz-Jeghers polyps or by finding even one such polyp in a patient with prominent mucocutaneous pigmentation.
8.2.3.2 Genetics
Peutz-Jeghers syndrome is an autosomal dominant hereditary disease, with incomplete penetrance (Harned et al. 1995). Approximately one quarter of cases appear to be due to new mutations (Gammon et al. 2009). Using currently available molecular genetic techniques, as many as 80–94 % of affected families have been found to carry defects in the STK11 gene (also known as LKB1), localizing to chromosome 19p13.3, with loss of heterozygosity suggesting function as a tumor suppressor gene (Hemminki 1999; van Lier et al. 2011). The majority are truncating or missense mutations, but up to 30 % may be large deletions, undetectable by sequencing alone (Aretz et al. 2005).
8.2.3.3 Clinical Manifestations
Clinically, patients have dark macules on the lips and buccal mucosa, occasionally on the eyelids, soles of the feet, and fingers. These mucocutaneous manifestations are benign, and are seen in all races and skin types, but are usually not visible in infancy. They have a tendency to fade with age, although the intraoral lesions are more persistent. Common symptoms related to the gastrointestinal polyps include abdominal pain, intestinal obstruction, and gastrointestinal bleeding. The disease may also present as intussusception during infancy and later in childhood (Harned et al. 1995; Howell et al. 1981). Approximately one third of patients are symptomatic during the first decade of life, and over half of patients become symptomatic before age 20 (Harned et al. 1995).
Peutz-Jeghers syndrome confers a significantly increased risk of malignancy both within and outside of the alimentary tract. Giardiello et al. found a relative risk of 15.2 for all cancers in Peutz-Jeghers syndrome, compared to the general population (Giardiello et al. 2000). In a recent study, van Lier et al. found a 76 % cumulative risk of developing cancer by the age of 70, with the risk greatest in female patients (van Lier et al. 2011). Although malignancy in Peutz-Jeghers syndrome usually manifests in younger and middle aged adults, children may also be affected. There is an increased incidence of both benign and malignant neoplasms, with breast cancer having the highest specific disease risk (45–54 %) (Gammon et al. 2009). Other associated neoplasms include pancreatic carcinoma, adenoma malignum (minimal deviation adenocarcinoma) of the uterus, testicular Sertoli cell tumor, and ovarian sex cord tumor with annular tubules (SCTAT). In fact, SCTAT are believed to occur in almost all female patients with Peutz-Jeghers syndrome. Isolated reports have been made of gallbladder carcinoma, bronchial adenoma, and biliary hamartomas (McGarrity et al. 2000).
Estimates of the frequency of gastrointestinal malignancy in Peutz-Jeghers syndrome have varied but are considerably less than in familial adenomatous polyposis (Harned et al. 1995; Flageole et al. 1994). In one recent study, the incidence of gastrointestinal carcinoma was 12 % by age 40, increasing to 51 % by age 70 (van Lier et al. 2011). Most investigators believe that there may be a progression from hamartomatous change to adenomatous change to carcinoma within some Peutz-Jeghers polyps, as foci of adenomatous change and adenocarcinoma have been identified in hamartomatous polyps from the stomach, small intestine, and colon (McGarrity et al. 2000; Narita et al. 1987; Matuchansky et al. 1979). Although the great majority of these gastrointestinal malignancies occur in adults, adolescents have been affected (Avendano-Garcia et al. 2002). Unusual gastrointestinal tumors have included a rectal carcinoid in an adolescent and an epithelioid stromal tumor arising from a hamartomatous polyp (Wada et al. 1998; Patterson and Kernen 1985).
8.2.3.4 Endoscopic Features
Polyps tend to occur in clusters and do not carpet the bowel. The polyps range in size and can get quite large, with most measuring between 0.8 and 5 cm. They can be sessile or pedunculated and cauliflower-like in gross appearance (Fig. 8.3) (Estrada and Spjut 1983).
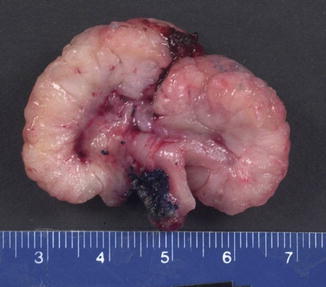
Fig. 8.3
Cut section of a polyp from the small intestine an 11-year-old boy with Peutz-Jeghers syndrome, demonstrating a pedunculated, cauliflower-like gross architecture
8.2.3.5 Pathology
Salient pathologic features of small bowel Peutz-Jeghers polyps are illustrated in Fig. 8.4. Microscopically, the polyps have an arborizing, treelike architecture, with prominent branching of thick smooth muscle bundles admixed with collagen within the lamina propria of the polyp. Although characteristic, the smooth muscle bundles are not pathognomonic for Peutz-Jeghers polyps, as they may also be seen in some pedunculated villous adenomas as well as in examples of mucosal prolapse syndromes (Fulcheri et al. 1991). The stroma may contain scattered lymphoid follicles, but the degree of inflammation is generally less than within juvenile polyps. The epithelium may be mitotically active and contain absorptive cells, goblet cells, argentaffin cells, and Paneth cells. As there is often only one epithelial cell type present in Peutz-Jeghers polyps of the stomach and colon, their hamartomatous nature may be difficult to appreciate. Pathologic variations encountered in Peutz-Jeghers polyps include enlarging mucinous cysts that can cause enteritis cystica profunda and epithelial misplacement within the frayed collagenous stroma of larger polyps (McGarrity et al. 2000). Pseudoinvasive changes, potentially mimicking invasive carcinoma, have been described in 10 % of small bowel Peutz-Jeghers polyps (Shepherd et al. 1987). These “misplaced” foci may penetrate beyond the submucosa and muscularis propria and even into the serosa but are usually devoid of dysplasia, and misdiagnosis of carcinoma can be avoided by the recognition of accompanying lamina propria with the misplaced glands (Bronner 2003).
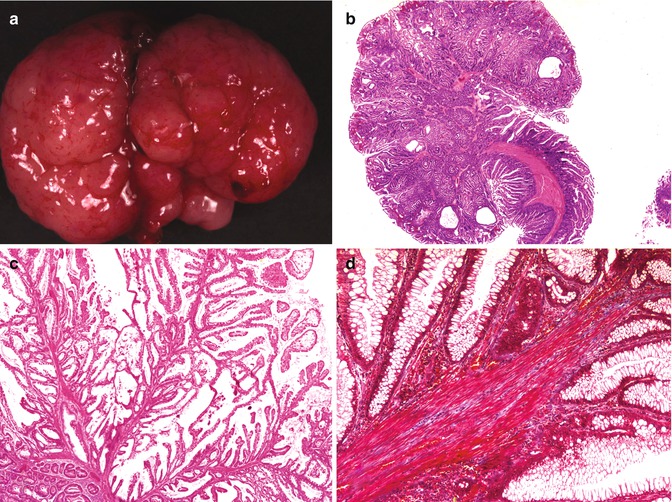
Fig. 8.4
Peutz-Jeghers polyps. (a) Pedunculated gastric Peutz-Jeghers polyp removed from a 6-year-old girl. (b) Histologic appearance of Peutz-Jeghers polyp of small intestine, with a prominent arborizing architecture. (c) Delicate frond-like growth typical of a Peutz-Jeghers polyp. (d) Prominent, brick-red smooth muscle fascicles within the stroma (Masson’s trichrome stain)
8.2.3.6 Treatment
In light of the risk of developing neoplasia, removal of all polyps greater than 1.5 cm in diameter is usually indicated (McGarrity et al. 2000). Although surveillance recommendations have been published, controlled trials of surveillance strategies are lacking in Peutz-Jeghers syndrome. The wide variety of neoplastic types and locations, as well as the age variability of presentation, raise unique challenges in following these patients. Esophagogastroduodenoscopy and colonoscopy, starting as early as age 10 and repeated at 2-year intervals, have been recommended by one group, as well as non-endoscopic surveillance via barium studies (McGarrity et al. 2000). Frequent interval screening for extraintestinal tumors is also of great importance, with particular emphasis on breast examination with mammography, as well as frequent testicular and pelvic examinations. As full discussion of the varied approaches to screening is beyond the scope of this chapter, the interested reader is referred to several references which discuss this in more detail (McGarrity et al. 2000; Giardiello et al. 2000; Hearle et al. 2006; van Lier et al. 2011).
8.2.4 PTEN Hamartoma Tumor Syndrome
8.2.4.1 Etiology
Cowden syndrome, or multiple hamartoma-neoplasia syndrome, is an uncommon genodermatosis characterized by hamartomas and neoplasms affecting all three germ cell layers in multiple organs (Harned et al. 1995). The estimated prevalence of Cowden syndrome is 1:200,000 to 1:250,000, and identification of patients has been facilitated by the development of diagnostic consensus criteria by the National Comprehensive Cancer Network (Gammon et al. 2009). Bannayan–Riley–Ruvalcaba syndrome (BRRS) encompasses what has previously been referred to in the literature as Ruvalcaba-Myhre-Smith syndrome, Bannayan-Zonana syndrome, and Riley-Smith syndrome and is a hamartomatous syndrome characterized by macrocephaly, lipomas, gastrointestinal polyposis, and pigmentary changes of the penis (Gammon et al. 2009; Parisi et al. 2001). Currently there are no consensus criteria for BRRS. Cowden syndrome and BRRS are closely related entities, as both are associated with PTEN gene mutations, and there is considerable clinical overlap between the two syndromes. Many investigators believe that PTEN mutation positive BRRS and Cowden syndrome represent different presentations of the same syndrome, collectively referred to as the PTEN hamartoma tumor syndrome (Marsh et al. 1999).
8.2.4.2 Genetics
Both Cowden syndrome and BRRS are transmitted as autosomal dominant conditions, associated with germ-line mutations of the PTEN gene, a tumor suppressor gene located on 10q22–q23 (Cantley and Neel 1999). Estimates of the frequency of these mutations vary in the literature, with figures as high as 80 % in patients with Cowden syndrome and as high as 60 % in patients with BRRS (Gammon et al. 2009). Other investigators have postulated a lower mutation frequency of 34 % in Cowden syndrome and have suggested the need for reevaluation of diagnostic criteria, which are complicated by the protean nature and variable phenotypic expression of these rare syndromes (Pilarski et al. 2011). In addition to mutations, a small number of Cowden syndrome patients have large deletions or rearrangements of the PTEN gene (Pilarski et al. 2011).
8.2.4.3 Clinical Manifestations
In addition to developing multiple hamartomatous polyps throughout the entire gastrointestinal tract, Cowden syndrome patients are prone to neoplasia of the skin, thyroid gland, kidney, uterus, and breast (Gorensek et al. 1984). Although the majority of cases are diagnosed in adults, young children can present with mental retardation, scrotal tongue, and progressive macrocephaly. Mucocutaneous signs such as tricholemmomas, fibromas, and palmer and plantar keratoses may become apparent in later childhood (Hanssen and Fryns 1995). Adult-onset Lhermitte-Duclos disease (cerebellar dysplastic gangliocytoma) is also part of the Cowden syndrome spectrum; presentation in childhood is less often associated with Cowden syndrome. BRRS is a congenital disorder, and patients exhibit a spectrum of lesions and clinical manifestations related to hamartomas including macrocephaly, mental retardation, pigmented penile macules, lipomas, hemangiomas, and gastrointestinal polyps. The most consistent feature of BRRS is megalencephaly without ventricular dilatation (Parisi et al. 2001).
8.2.4.4 Pathology
Gastrointestinal polyps in Cowden syndrome are often asymptomatic and have heterogenous morphologic features, with many having combined features of both juvenile and hamartomatous polyps. Indeed, since the polyps of Cowden syndrome are not histologically pathognomonic, it has been suggested that the presence of multiple polyps of varied histologic appearance should raise a suspicion for Cowden syndrome (Stanich et al. 2011). The polyps are present in at least 40–70 % of patients and can develop anywhere from the mouth to the anus. Common sites include the stomach, colon, esophagus, and duodenum (Kato et al. 2000). Although similar to Peutz-Jeghers polyps, these polyps are more often sessile and tend to be smaller, less exophytic, and less arborizing. In addition, these patients also develop lipomatous, hyperplastic, ganglioneuromatous, and inflammatory polyps. Polyps occurring in BRRS usually occur in the colon and have the features of typical juvenile polyps (Lowichik et al. 2000).
8.2.4.5 Treatment
Although adult-onset colorectal carcinoma has been reported in Cowden syndrome, the significance of this has not been established. Likewise, although there have been reports of low-grade adenomatous changes in the juvenile polyps of BRRS, there have been no reports to date of adenocarcinoma developing in these polyps. Treatment guidelines for the heightened risk for extraintestinal malignancy are available and are beyond the scope of this chapter. As the overall risk of gastrointestinal malignancy is uncertain and appears to mainly to manifest in adulthood, there are no established recommendations for routine gastrointestinal screening in childhood.
8.2.5 Tuberous Sclerosis and Proteus Syndrome
Hamartomatous polyps, histologically identical to Peutz-Jeghers polyps have been described in tuberous sclerosis. In one series, 75 % of patients with the genodermatosis were found to have colonic polyps, which were either hamartomatous, adenomatous, or mixed (Devroede et al. 1988). Juvenile polyps of the rectum have also been described in Proteus syndrome, a rare congenital hamartomatous disorder associated with hemihypertrophy, gigantism of the extremities, nevi, angiomas, and lipomas (Lamireau et al. 1993).
8.2.6 Cronkhite-Canada Syndrome
Essentially a syndrome affecting adults, juvenile polyps occur throughout the gastrointestinal tract, with hyperplastic morphology in the stomach. Most polyps are sessile, but otherwise these polyps lack unique morphologic features. The condition is noninherited and is associated with alopecia, onychodystrophy, and macular skin lesions. Severe malabsorption is present in most patients and is associated with protein-losing enteropathy and malnutrition. The terms “juvenile” or “infantile” Cronkhite-Canada syndrome have been used in some case reports of juvenile polyposis coli associated with hypoproteinemia. However, in the absence of the ectodermal changes seen in adults, it has been recommended that these cases be considered as expressions of juvenile polyposis, distinct from adult Cronkhite-Canada syndrome (Burke and Sobin 1989).
8.2.7 Adenomatous Polyps and Familial Adenomatous Polyposis
8.2.7.1 Etiology
Sporadic adenomatous polyps are generally found in middle aged and older adults and are rare in children. The presence of even one adenomatous polyp in a child should raise a suspicion for a polyposis syndrome, as it may herald the future development of many more polyps (Winter 2000). Less than one percent of colorectal polyps in children in one series were adenomatous, and of these more than half occurred in patients with syndromic polyposis (Louw 1972). As these polyps are usually encountered in children with adenomatous polyposis syndromes, the pathologic features will be discussed in the context of these syndromes.
Familial adenomatous polyposis (FAP) is an autosomal dominant inherited disorder in which affected family members develop numerous colorectal adenomatous polyps, with a virtually 100 % risk of developing invasive colorectal carcinoma before reaching middle age, unless the colon is prophylactically removed. The disease has an incidence of 1 in 8,000, which is maintained by a frequency of new mutations, accounting for approximately 25 % of cases (Fearnhead et al. 2001). Variants of FAP include Gardner’s syndrome and Turcot’s syndrome. Although polyps are rare in early childhood, they usually become endoscopically visible by adolescence or early in the third decade of life. Previously, the disease was referred to as familial polyposis coli or adenomatous polyposis coli, terms now replaced by familial adenomatous polyposis (FAP), acknowledging the fact that polyps commonly occur in the stomach and small bowel as well as the colon.
Gardner’s syndrome is a variant of FAP, characterized clinically by the development of osteomas and varied soft tissue tumors, including epidermal cysts, lipomas, fibromas, and desmoid tumors, in addition to intestinal adenomatous polyposis. Desmoid tumors develop in 10 % of patients with FAP, often following abdominal surgery and can also occur in childhood, preceding development of adenomatous polyps (Clark et al. 1997). Fundic glandular cysts also occur, and the risk for colorectal carcinoma is unchanged from FAP in general (Eichenberger et al. 1980). The APC gene is mutated in Gardner’s syndrome but at a different location than in FAP unassociated with desmoids.
Turcot’s syndrome refers to intestinal polyposis coexisting with tumors of the central nervous system. In some cases, patients with APC gene mutations develop brain tumors (usually medulloblastoma) as part of the spectrum of FAP. However, in other cases, also referred to as Turcot’s syndrome, patients with mismatch repair gene mutations (typical of hereditary nonpolyposis colon cancer, or HNPCC) also develop brain tumors, usually glioblastoma multiforme (Hamilton et al. 1995).
8.2.7.2 Genetics
FAP is caused by a mutation of the APC (adenomatous polyposis coli) gene located at 5q21, and germ-line mutations achieve a near-100 % penetrance (Foulkes 1995). The APC gene plays a key role in epithelial cell homeostasis, being involved in cell cycle progression, migration, differentiation, and apoptosis. It functions as a tumor suppressor gene, encoding a protein which associates with β-catenin, preventing its role in cellular proliferation. When mutated, the truncated abnormally functioning protein does not properly bind (Fearnhead et al. 2001). In accordance with Knudson’s two hit model of carcinogenesis, colorectal cancers in FAP patients have additional somatic APC mutations or loss of heterozygosity, in addition to the germ-line APC mutation. To date, over 900 distinct mutations have been identified within the 15 exons at the APC locus in patients with FAP (Alkhouri et al. 2010; Hernegger et al. 2002). Approximately 95 % of these mutations give rise to truncated APC protein (Macrae et al. 2009). New FAP mutations frequently are associated with a severe phenotype and the site of mutation may predict the severity and onset of the disease (Gayther et al. 1994; Caspari et al. 1994). In addition to its role in FAP, APC mutations have been discovered in up to 80 % of all colon cancers and adenomas (Goss and Groden 2000).
It has been suggested that asymptomatic at risk individuals for FAP or AFAP undergo genetic testing between ages 10 and 15 to determine the presence of an APC germ-line mutation. Routine mutation detection techniques disclose germ-line APC mutations in up to 70 % of individuals with classic FAP. More complex genetic testing may be necessary in individuals with suspected AFAP and individuals in whom FAP is suspected, but routine mutation techniques are negative (Sieber et al. 2002). Such techniques may include linkage analysis, direct gene sequencing, and protein truncation testing (Macrae et al. 2009). If a child in a family with a specific identified mutant APC allele is found to be mutation negative, that child has the same risk of developing colonic carcinoma as the general population (Alkhouri et al. 2010).
8.2.7.3 Clinical Manifestations
In addition to the development of colorectal adenomas and carcinoma, FAP is associated with a generous number of extracolonic lesions, both neoplastic and hyperplastic, which are summarized in Table 8.4. Among these are congenital hypertrophy of the retinal pigment epithelium, development of which is a diagnostically useful sign in children born into FAP kindreds. Hepatic tumors have also been described, consisting of hepatoblastomas (Garber et al. 1988; Hughes and Michels 1992; Kingston et al. 1983; Li et al. 1987), hepatic adenomas, and neoplastic adenoma-like nodules, possibly representing well-differentiated hepatocellular carcinoma (de Chadarevian et al. 2002). FAP patients have a 25-fold increased incidence of nasopharyngeal angiofibroma, a rare tumor (Giardiello et al. 1993). Mesenteric fibromatosis, often associated with “Gardner’s-like” signs has been described in 13 % of FAP patients (Lotfi et al. 1989). Other tumors that have been reported include rhabdomyosarcoma, papillary thyroid cancer, and adrenocortical tumors (Fearnhead et al. 2001).
Table 8.4
Extracolonic manifestations of familial adenomatous polyposis
Adenomas |
Adrenal |
Hepatic |
Pituitary |
Small bowel |
Gastric |
Carcinomas |
Adrenal |
Thyroid |
Breast |
Gastric |
Bile duct/periampullary/pancreatic |
Small bowel |
Other malignancies |
Carcinoids |
Brain tumors (Turcot’s syndrome) |
Hepatoblastoma |
Rhabdomyosarcoma |
Other lesions |
Osteomas |
Gardner’s associated fibromatosis |
Lipomas |
Nasopharyngeal angiofibroma |
Desmoids |
Epidermal cysts |
Impacted and supernumerary teeth |
Congenital hypertrophy of the retinal pigment epithelium |
Fundic gland polyps and cysts |
Gastroduodenal pathology is present in most patients with FAP. In one series, 60 % of FAP patients had gastric polyps, and 70 % had duodenal polyps (Goedde et al. 1992). The gastric polyps in FAP usually are fundic gland polyps manifesting as small excrescences in the body and fundus. They occur both in FAP and also in attenuated familial polyposis (see below), where they are a near-universal finding. Although generally considered to be benign lesions, there is a report of gastric adenocarcinoma occurring in an 11-year-old girl in the setting of AFAP and fundic gland polyps with severe dysplasia (Attard et al. 2001). Not pathognomonic for FAP, fundic gland polyps may also be seen as sporadic lesions, usually at an older age. However, in children, fundic gland polyps are strongly associated with FAP (Attard et al. 2002).
Between 50 and 90 % of patients with FAP develop visible duodenal adenomas, usually around the major papillae (Domizio et al. 1990). In patients who have received prophylactic colectomies, duodenal carcinomas rank with desmoid tumors as a major cause of death.
In a subset of patients with attenuated familial adenomatous polyposis (AFAP), fewer than 100 colorectal polyps develop. Patients with AFAP tend to be older when they develop both adenomas and carcinoma and are less likely to present as children. Desmoids and osteomas are uncommon in these patients.
8.2.7.4 Endoscopic Features
Colonic adenomas are not present at birth and are extremely rare in early childhood. Visual polyps typically appear around the time of puberty, and by the early third decade, the colon is typically carpeted by hundreds or thousands of adenomas, although there is considerable variation depending upon the severity of the disease and the age of the patient (Fig. 8.5). No portion of the colon is spared, and most of the polyps are small, less than 0.5 cm in diameter, although some can become quite large. Smaller colonic polyps tend to be sessile, whereas the larger ones may be either sessile or pedunculated.
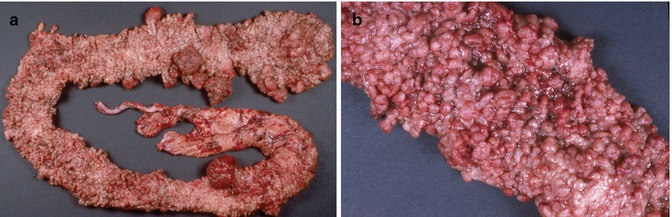
Fig. 8.5
(a) Colectomy specimen, 19-year-old girl with familial adenomatous polyposis, illustrating innumerable polyps throughout the length of the specimen. Some of the polyps have attained considerable size. (b) Close-up view demonstrating diffuse polypoid transformation of the colonic mucosa
8.2.7.5 Pathology
In the colon, histologic abnormalities can be found in both polypoid and grossly unremarkable mucosa. The earliest microscopic changes in FAP consist of tiny areas of mild dysplasia, in which small groups of adenomatous tubules can be found adjacent to normal appearing glands (Fig. 8.6). These changes may be limited to a single or several crypts, and when such focal changes are seen in a biopsy, the diagnosis of FAP must be strongly considered. As the lesions evolve, they assume gross and microscopic appearances similar to those of sporadic tubular, tubulovillous, and villous adenomas, with various degrees of dysplasia (Fig. 8.7). Although invasive colonic adenocarcinoma is unusual in childhood, a recent review of colectomy specimens from children aged 18 and younger found evidence of severe dysplasia or carcinoma in situ in 27 % (Vasudevan et al. 2006).
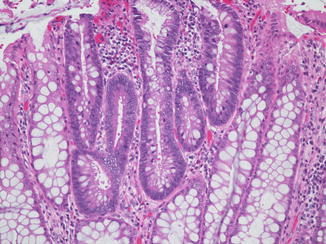
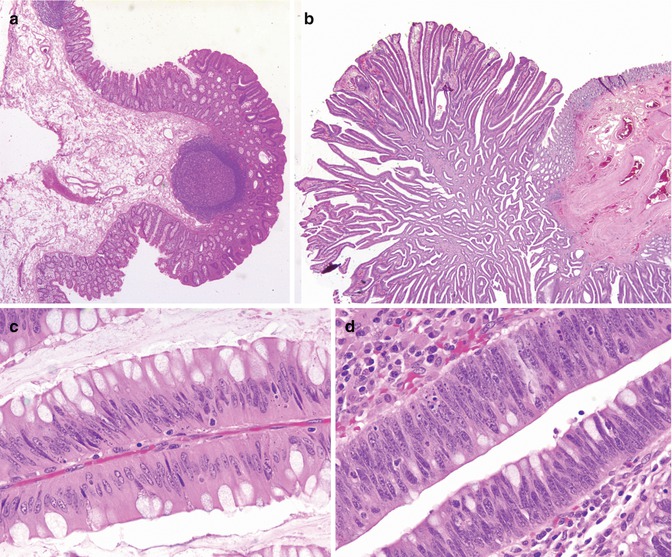
Fig. 8.6
Adenomatous changes within the colonic mucosa of a patient with familial adenomatous polyposis. Several non-transformed crypts remain at the periphery of the photomicrograph
Fig. 8.7
Familial adenomatous polyposis, colon. (a) Tubular adenoma. (b) Villous adenoma. (c) Mild dysplasia. (d) Severe dysplasia
Fundic gland polyps are most likely hamartomatous in nature, histologically composed of disordered and tortuous gastric fundic glands containing parietal chief and mucus neck cells, with cystic glandular dilatations extending near to the surface (Fig. 8.8).
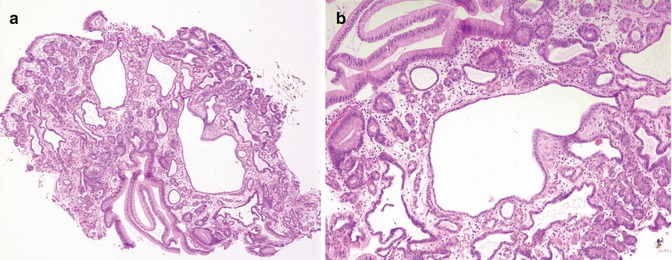
Fig. 8.8
(a) Low-power histology of fundic gland polyp removed from a 13-year-old girl with Gardner’s syndrome. (b) Higher-power photomicrograph demonstrating disordered and cystic fundic glands
8.2.7.6 Treatment
In the pediatric FAP population, colectomies are usually performed prophylactically but are often delayed until adolescence. The risk of developing cancer in children less than 21 years of age has been estimated to be as low as 0.21 % to as high as 7 % (Alkhouri et al. 2010). The definitive treatment to avoid the certain development of colorectal carcinoma in FAP is total colectomy with proctectomy and ileoanal anastomosis, usually performed in late adolescence or early in the third decade of life. Preceding this, specific recommendations vary, although it is generally agreed that regular endoscopic surveillance with biopsies should commence early in the second decade at the latest. Interval esophagogastroduodenoscopy to evaluate the stomach and duodenum is recommended by age 20–25 (with some advocating for earlier evaluation) (Alkhouri et al. 2010). The necessity for surveillance of the small bowel distal to the duodenum is unsettled, as the development of carcinoma in the jejunum and ileum is unusual in FAP (Alkhouri et al. 2010). The pathologist should be prepared to carefully evaluate all polyps and mucosal biopsies from these patients for evidence of adenomatous change and dysplasia. In addition, alpha-fetoprotein serum screening has been recommended for children at risk for FAP, given the higher risk of hepatoblastoma (Attard et al. 2001).
8.2.8 Hyperplastic Polyps
Hyperplastic polyps are occasionally found in children. Although one of the most common polyps in the adult colon, there is a paucity of literature on these polyps in children, as they seldom assume clinical significance. In general, hyperplastic polyps are smaller than their adenomatous and hamartomatous counterparts, most commonly assuming a sessile profile. Histologically, hyperplastic polyps are composed of groups of elongated, hyperplastic crypts, resulting in mucosal thickening, exaggerated crypt architecture, and often a “sawtooth” or serrated epithelial architecture imposed by papillary infolding of the crypt epithelium (Fig. 8.9).
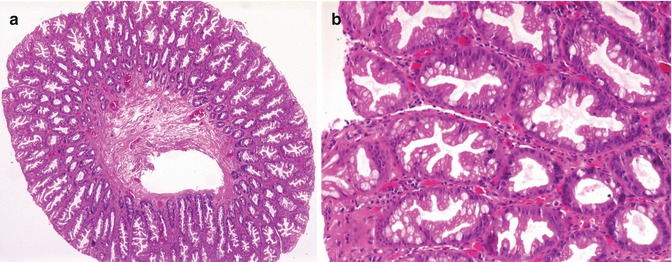
Fig. 8.9
(a) Low-power microscopic appearance of hyperplastic colonic polyp. (b) Higher power photomicrograph demonstrating the characteristic “saw-tooth” epithelial architecture
The region of the esophagogastric junction is the site of hyperplastic and inflammatory polyps that are believed in many cases to be related to reflux disease or some form of esophageal injury. Although these hyperplastic polyps are usually seen in adults, they may occur in children (Abraham et al. 2001). Histologic examination reveals hyperplastic epithelium (foveolar, squamous, or combined) with a variably inflamed stroma. Most are completely or nearly completely foveolar, with a minority being predominantly squamous; associated severe esophagitis, sometimes with strictures, is commonly seen. These polyps have also been described in children with NF1 (De Giacomo et al. 1994) and have been associated with gastric outlet obstruction in neonates (Brooks et al. 1992; Sanna et al. 1991).
8.2.9 Inflammatory Polyps
The majority of “inflammatory polyps” seen in children and indeed also in adults are pseudopolyps, in the sense that they do not represent true mucosal proliferations, but rather are admixtures of hyperplastic and inflamed mucosa, arising in the setting of intense inflammation, most commonly secondary to inflammatory bowel disease or infectious colitis. Distinctive types of inflammatory polyps include inflammatory fibroid polyps, inflammatory cloacogenic polyps, and pyogenic granulomas.
Although inflammatory pseudopolyps may grossly resemble adenomatous polyps and can attain substantial size, they have no inherent malignant potential. In addition to occurring in the setting of inflammatory bowel disease and infection, they also commonly occur at sites of mucosal injury, such as at surgical anastomoses, and in areas of stricture secondary to necrotizing enterocolitis (Iofel et al. 2000). Large pseudopolyps can cause intestinal obstruction (Adelson et al. 1988). Histologically, these pseudopolyps are composed of hyperplastic, often ulcerated mucosa, with subjacent intensely inflamed and edematous submucosa and granulation tissue. The marked hyperplastic and regenerative epithelial changes may mimic dysplasia.
A closely related entity is the inflammatory polyp–fold complex occurring at the gastroesophageal junction. As with hyperplastic polyps in this location, this lesion is usually seen in the setting of reflux esophagitis. Endoscopically, a round firm lesion is visualized at the gastroesophageal junction, often in continuity with a prominent gastric mucosal fold (Bishop et al. 2002). The histology is nonspecific, with components of benign esophageal and gastric mucosa and a mixed inflammatory infiltrate. Although more commonly seen in adults, this lesion has been described in children (Bishop et al. 2002; Kato et al. 1993).
Inflammatory fibroid polyps are relatively uncommon lesions. Essentially polypoid excrescences of granulation tissue, which are larger than pyogenic granulomas, have been variously labeled in the past as inflammatory pseudotumors, submucosal fibromas, and “Vanek” polyps. No underlying cause or medical condition has been associated with these polyps. In a series of 64 patients, one pediatric case (a 3-year-old) was described, and isolated case reports exist of this lesion in children, including a rectal inflammatory fibroid polyp in an 8-year-old boy (Shimer and Helwig 1984; Pollice and Bufo 1984). In all ages, these lesions have been benign, although usually of large size, ranging up to 13 cm in diameter, with most 2.6–5.0 cm in diameter. The polyps are commonly ulcerated and are composed of a stroma rich in fibroblasts, granulation tissue-like capillaries, and a diffuse inflammatory infiltrate of eosinophils, lymphocytes, macrophages, plasma cells, and mast cells. Hemosiderin is commonly seen. The most common site of the lesion is the stomach, followed by the small bowel.
Inflammatory cloacogenic polyp (ICP) is an inflammatory polyp that arises in the anorectal transition zone and clinically and pathologically has similarities to solitary rectal ulcer syndrome (SRUS). Although usually described in adults, the lesion is not rare in children, as there are several case reports and small series of pediatric patients, most of whom have been school-aged or adolescents (Washington et al. 1993; Saul 1987; Poon et al. 1997; Bass et al. 1995). Patients with ICP typically present with hematochezia and tenesmus; there may be a preexisting history of SRUS, Crohn’s disease, or rectal prolapse. Histologically these polyps have a tubulovillous architecture and contain both stratified squamous and columnar mucosa in variable proportions (Fig. 8.10). Many contain cloacal-type mucosa, often juxtaposed with areas of ulceration and granulation tissue (Saul 1987). Commonly there is disorganization of the smooth muscle of the muscularis mucosa, with extension of smooth muscle bundles into a somewhat fibrotic lamina propria, suggesting a etiologic role for mucosal prolapse, ischemia, and a link with SRUS (Poon et al. 1997). These polyps may recur following polypectomy.
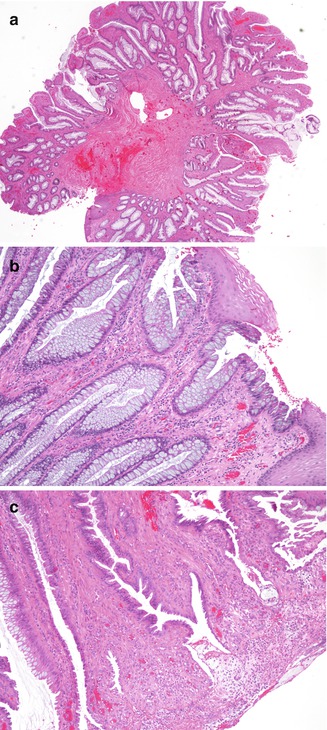
Fig. 8.10
Histologic appearances of inflammatory cloacogenic polyp. (a) Low-power photomicrograph of polyp removed from rectum of a 17-year-old boy. (b) High-power photomicrograph shows presence of both squamous and colonic epithelium. (c) Another high-power image demonstrating prominent smooth muscle within the lamina propria and surface ulceration and inflammation
Pyogenic granulomas in the gastrointestinal tract are similar histologically to those occurring elsewhere in the body. Discriminating these from small or incipient juvenile polyps with abundant inflammation and ulceration can be difficult and may not always be possible.
8.2.10 Lymphoid Polyps
Aggregates of lymphoid tissue may give rise to polyps, in children most commonly in the rectum but also occasionally elsewhere in the alimentary tract (Byrne et al. 1982; Atwell et al. 1985; McNicholas et al. 1985; Rutsch et al. 1997). Lymphoid polyps are usually small, sessile excrescences, ranging from 0.1 to 0.5 cm, and may be centrally umbilicated (Gonzalez-Peralta and Andres 1999). They can be multiple and rarely can present as a single larger mass up to 5 cm in diameter. Histologically, the polyps contain reactive lymphoid tissue with germinal centers (Fig. 8.11). In addition to intussusception, complications include rectal prolapse, intestinal pseudoobstruction, rectal bleeding, and abdominal discomfort.
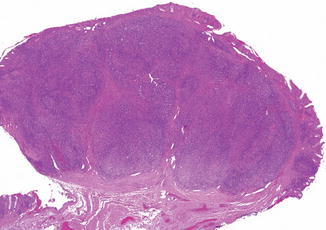
Fig. 8.11
Rectal lymphoid polyp
In contrast to lymphoid polyps, lymphoid hyperplasia is commonly seen in the pediatric intestines, generally most prominent in the terminal ileum, where the submucosa is often subtotally replaced by follicular lymphoid tissue. In this location, the histology is so common as to suggest a normal response to antigenic stimulation in children.
8.3 Tumors of the Gastrointestinal Tract
A wide variety of tumors can affect the alimentary tract in children. Favored locations of both common and uncommon tumors are summarized in Table 8.5.
Table 8.5
Favored locations of selected gastrointestinal tumors in children
Histologic type | Usual location |
|---|---|
Epithelial | |
Squamous cell papilloma | Esophagus |
Squamous cell carcinoma | Esophagus |
Adenocarcinoma | Colon (occ. stomach, esophagus) |
Carcinoid | Appendix |
Mesenchymal | |
Lymphangioma | Mesentery |
Leiomyoma | Esophagus, rectum |
Gastrointestinal stromal tumor | Stomach, small bowel |
Inflammatory myofibroblastic tumor | Mesentery |
Ganglioneuroma | Colon, rectum |
Rhabdomyosarcoma | Perianal region |
Desmoplastic small round cell tumor | Mesentery |
Lymphoma | Small bowel |
Teratoma | Stomach |
8.3.1 Nonneoplastic Conditions
Nonneoplastic conditions of the gastrointestinal tract and mesentery can present clinically as a mass, prompting excision or biopsy to rule out neoplasia. Inflammatory conditions such as ileocecal Crohn’s disease, mesenteric lymphadenitis (Jelloul et al. 1997; Koruda et al. 1988), intestinal amebiasis (Ciftci et al. 1999), and perianal abscesses can mimic neoplasms in their presentation, as can malformative lesions such as enteric duplications and heterotopias, which will be covered elsewhere in this volume. Hamartomatous and choristomatous lesions in childhood are among the most frequent developmental anomalies of the pediatric GI tract to mimic neoplasia. By strict definition, hamartomas contain disordered tissues native to the organ involved, while choristomas contain foreign tissues. Depending upon one’s definition of whether the gastrointestinal tract fulfills the requirements of an “organ,” the distinction between the two in the literature may be somewhat blurred.
Fibromuscular hamartoma of the esophagus, clinically mimicking a neoplasm, was described in an 11-month-old infant (Beckerman et al. 1980), and rectosigmoid hamartoma associated with rectal prolapse has been seen in a 14-week-old infant (Lamesch 1983). Omental-mesenteric-myxoid hamartoma is a rare lesion, first described in three infants by Gonzalez-Crussi et al. in 1983, characterized by multiple tumoral nodules of the omentum and mesentery, histologically composed of plump mesenchymal cells in a myxoid, well-vascularized stroma (Gonzalez-Crussi et al. 1983). Highly cellular, the infantile lesion can mimic a spindle-cell sarcoma and has similarities to the inflammatory myofibroblastic tumor (IMT) yet is most likely of a hamartomatous nature with a favorable prognosis. Brunner’s gland hamartomas, composed of a predominance of histologically normal Brunner’s glands, can take the form of polypoid or mass lesions and precipitate pyloric obstruction, gastrointestinal hemorrhage, and intussusception in children (Hedges 1988). A related lesion, possibly overlapping pathophysiologically with Brunner’s gland hamartoma, is the adenomyoma which may contain a variety of epithelial tissues (ductal structures, Brunner’s glands, exocrine and endocrine pancreatic tissues) as well as smooth muscle (Chan and Roche 1994; Stewart and Mills 1984). Pathologic examination reveals a polypoid mass, composed of cystically dilated glands lined by tall columnar epithelium resembling hepatobiliary or pancreatic ductal epithelium, hypertrophic smooth muscle bundles, and fibrous stroma (Kim et al. 1990). In children the lesion is most commonly seen in the gastric antrum, although it has been described in the proximal small bowel, and rarely in the ileum, where it has been associated with intussusception (Kim et al. 1990; Serour et al. 1994).
8.3.2 Epithelial Neoplasms of the Esophagus
The most common epithelial neoplasm of the esophagus in children is the squamous cell papilloma. These lesions are similar histologically to those seen in the larynx, and the majority occur in the lower third of the esophagus (Fig. 8.12) (Polit 1990). In one study, 50 % of esophageal papillomas contained HPV genetic material (Odze et al. 1993). The esophagus can also be involved by respiratory papillomatosis (Batra et al. 2001).
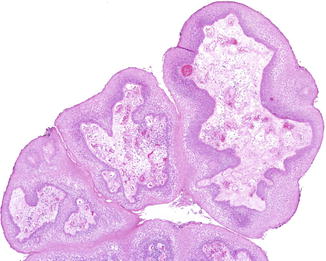
Fig. 8.12
Squamous cell papilloma, esophagus
Twenty-four cases of esophageal carcinoma occurring in children ranging from 8 to 20 years old have been reported to date (Issaivanan et al. 2012; Al Hilou et al. 1984; Schettini et al. 1998; Gangopadhyay et al. 1997; Zotter et al. 2001). 60 % have been adenocarcinomas, suggesting that many of these tumors developed in a background of Barrett’s esophagitis. This relationship in adolescence was documented by Hassall et al. in a report of a 17-year-old boy with adenocarcinoma and multifocal high-grade dysplasia arising in a background of Barrett’s esophagitis. This report cites nine other cases in the literature of Barrett’s related adenocarcinoma developing between ages 11 and 25 (Hassall et al. 1993). Caustic ingestion is also a risk factor for both pediatric adenocarcinoma and squamous cell carcinoma. An exceptional case of squamous cell carcinoma occurred in an esophageal duplication cyst (Singh et al. 2001), and another reported case of childhood esophageal carcinoma arose 10 years after acid ingestion injury (Schettini et al. 1998). As in adults, the overall prognosis of esophageal carcinoma is poor in children (Issaivanan et al. 2012).
8.3.3 Epithelial Neoplasms of the Stomach
Although gastric carcinoma is rare in childhood, there are numerous individual case reports of its occurrence (Bachmeyer et al. 2000; Conley et al. 1988; Chatura et al. 1996; Katz et al. 1997; Michalek et al. 2000). Its rarity is attested to by the fact that in an 18-year period, only five cases of pediatric gastric adenocarcinoma were diagnosed at the University of Texas M.D. Anderson Cancer Center, and in a 25-year period, only two cases were diagnosed at the Children’s Hospital of Los Angeles (Subbiah et al. 2011; Mahour et al. 1980). Most pediatric cases are diagnosed in teenagers. Pediatric gastric adenocarcinoma has been associated with ataxia telangiectasia (Siegel et al. 1976), common variable immunodeficiency (Conley et al. 1988), dyskeratosis congenita (Chatura et al. 1996), X-linked agammaglobulinemia (Bachmeyer et al. 2000), and atrophic gastritis (Katz et al. 1997). Both anaplastic infiltrative and mucinous forms occur. As in adults, the overall prognosis is poor.
Gastroblastoma is a rare and newly described biphasic epitheliomesenchymal stomach tumor, first described in a series of three young adults (Miettinen et al. 2009a). A single case has been reported in a 9-year-old boy (Shin et al. 2010). This tumor is composed of a mesenchymal component of spindle to oval cells without significant atypia and an epithelial component consisting of either of clusters of epithelioid cells or luminal structures with inspissated secretions. The epithelial components stain with keratins and the mesenchymal components with CD10 and vimentin. The reported clinical behavior of these very rare neoplasms has been excellent following treatment.
8.3.4 Epithelial Neoplasms of the Small Bowel
8.3.5 Colorectal Carcinoma
8.3.5.1 Etiology
Although uncommon as compared to adults, colorectal carcinoma in the pediatric age group is not extraordinarily rare, as the literature is replete with both single case reports and larger series. In fact there are at least six separate large series in the literature (more than 10 patients per series), summarized in Table 8.6, in which the number of included patients ranges from 29 to 159, totaling 399 cases (Hill et al. 2007; Sultan et al. 2010; Andersson and Bergdahl 1976; Buyukpamukcu et al. 1996a; LaQuaglia et al. 1992; Yamamoto et al. 1994). However, as some of these series were from large databases, it is likely that some patients may have been counted in more than one series. Approximately 80 new cases per year are reported in the United States, with an overall incidence estimated to be 1 in 10 million (Blumer et al. 2012). Although most cases are described in adolescents, the tumor has occurred in infants and very young children. Most, but not all, children and adolescents who develop colon cancer have either FAP or hereditary nonpolyposis colorectal cancer syndrome (HNPCC).
Table 8.6
Selected large series of colorectal cancer in children
Series | Age range | Number of cases | Comments |
|---|---|---|---|
Andersson and Bergdahl (1976) | Childhood | 75 | 5-year survival 2.5 % |
General Literature Review | |||
LaQuaglia et al. (1992) | 10–21 | 29 | 45 % signet ring histology |
Memorial Sloan-Kettering Cancer Center | 76 % lymph node metastases at presentation | ||
Median survival: 16 months | |||
Yamamoto et al. (1994) | <15 | 43 | Majority of patients older than 10 |
Japanese Literature Review | |||
Buyukpamukcu et al. (1996a) | Adolescents | 16 | 75 % mucinous histology |
Turkey (Hacettepe University Children’s Hospital) | 1 patient known to be alive after 4 years | ||
Hill et al. (2007) | 7–19 | 77 | 86 % advanced stage disease (stage 3 or 4) |
St. Jude Children’s Research Hospital | 62 % mucinous histology | ||
20 % survival at 10 years | |||
Sultan et al. (2010) | 4–20 | 159 | 22 % mucinous histology |
SEER database 1973–2005 (United States) | 18 % signet ring histology | ||
40 % survival at 5 years |
Unusual associations described in single case reports of children include IgA deficiency (Hamoudi et al. 1974) and tuberous sclerosis (Digoy et al. 2000). Colonic adenocarcinoma can occur as a complication of ureterosigmoidostomy performed for bladder exstrophy. Cancer has developed between 10 and up to 46 years after the procedure (Eraklis and Folkman 1978) and may be preceded by evidence of dysplasia (Fig. 8.13). Colorectal carcinoma has also occurred in children as a second malignancy following irradiation for childhood tumors.
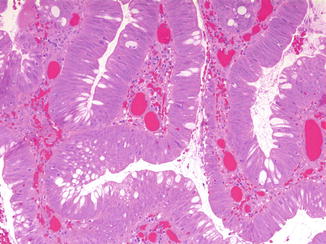
Fig. 8.13
Severe dysplasia arising in polypoid colonic mucosa at ureterosigmoidostomy site in a 20-year-old woman with congenital bladder exstrophy
Colorectal carcinoma is a well-known complication of inflammatory bowel disease. Although aneuploidy and dysplasia have been found in patients as young as 16 years old with inflammatory bowel disease, frank carcinoma in the setting of IBD is unusual in children and adolescents (Markowitz et al. 1997). Nevertheless, given estimates that approximately 10 % of patients with ulcerative colitis develop colorectal carcinoma for every decade after disease activity has started, it would be prudent to carefully monitor teenagers who developed inflammatory bowel disease in early childhood (Blumer et al. 2012).
8.3.5.2 Genetics
As a major cause of adult mortality, colorectal carcinoma has been the subject of intense molecular genetic scrutiny, both in defining underlying genetic susceptibility and in unraveling somatic genetic defects present in these cancers. However, given its relative rarity in childhood, there is little specific molecular genetic data available for pediatric disease. It is estimated that 15–30 % of all colorectal carcinomas, regardless of age, have a major hereditary component, and the bulk of these cases are due to either the hereditary nonpolyposis colorectal cancer syndrome (HNPCC) or familial adenomatous polyposis (FAP) (Fearon 2011). The genetics underlying FAP were discussed earlier in this chapter. As with FAP, HNPCC is inherited in an autosomal dominant fashion, but unlike FAP, these patients lack mutations of APC. Instead, many HNPCC patients have loss of function of the tumor suppressor genes MLH1 and MSH2 (Aaltonen 2000).
HNPCC in particular, even more so than FAP, is associated with the development of colorectal carcinoma at an unusually young age. HNPCC also carries an increased lifetime risk of cancers of the endometrium, stomach, biliary tract, urinary tract, and ovaries (Aarnio et al. 1995). Although most cancers occur in early middle age, colorectal adenocarcinoma has been described in young teenagers with HNPCC (Lynch et al. 1991; Madlensky et al. 1997).
8.3.5.3 Clinical Manifestations
Most patients present with pain, altered bowel habits, and/or weight loss (Hill et al. 2007). Additional common symptoms may include vomiting, constipation, diarrhea, and rectal bleeding; anemia is also common. In addition the vast majority of pediatric patients presenting with colorectal carcinoma do not carry a previously established diagnosis of FAP.
Although the clinical presentation of colorectal carcinoma is similar in children and adults, pediatric colorectal carcinoma has a tendency toward higher stage at diagnosis than its adult counterpart (Angel et al. 1992). In one series, the median survival rate of all patients was only 16 months, and 76 % of children had lymph node metastases at presentation (LaQuaglia et al. 1992). In a large series from St. Jude Children’s Research Hospital, 86 % of patients presented with advanced stage (stage 3 or 4) disease at diagnosis, with only a 20 % 10-year survival (Hill et al. 2007). In a recent review of the US SEER database, the 5-year overall survival for pediatric colorectal carcinoma was only 40 % (Sultan et al. 2010). Although delayed diagnosis of an unexpected disease in children may play a role in the overall poor prognosis, it seems clear that pediatric colon cancer is in general a biologically more aggressive malignancy than its adult counterpart.
8.3.5.4 Radiographic Features
Abdominal and pelvic CT and MRI can be used for initial evaluation, to determine disease extent and to evaluate for recurrence. Common imaging findings include polypoid primary tumors and “apple core” regions of colonic stenosis. These examinations typically will reveal a homogenously and diffusely low attenuation mass involving a segment of colon, with circumferential or partial circumferential involvement, in the absence of free fluid or mesenteric inflammation (Blumer et al. 2012). Cystic, necrotic, and hemorrhagic areas may be present. The disease may occur anywhere in the colon, with an approximately equal distribution between the right and left sides. Advanced disease may be heralded by regional adenopathy or liver metastases.
8.3.5.5 Pathology
Given that excellent and exhaustive descriptions of the pathologic features of colorectal cancer are available, this chapter will instead focus on features typical of the disease in childhood. Grossly, pediatric colonic carcinomas resemble their adult counterparts, although they are more likely to have the gelatinous appearance of the mucinous subtype. Histologically, over half, and in some series, over 75 % of childhood colon cancers are poorly differentiated mucinous tumors (Fig. 8.14) (Odone et al. 1982; Rao et al. 1985; Chabalko and Fraumeni 1975). This contrasts with adults, in whom over 90 % of cases are moderately to well-differentiated adenocarcinomas. Mucinous carcinomas have a significantly poorer prognosis than ordinary adenocarcinomas, with a greater tendency to invade contiguous structures, to develop distant lymph node metastases, and to recur following surgical resection. Mucinous carcinomas (composed of at least 25–50 % mucinous histology) of both the colloid and signet ring types are increased in children. Colloid carcinoma is characterized by abundant pools of extracellular mucin associated with malignant glandular epithelium. In colloid carcinomas, the extracellular mucin may be so extensive, especially in metastatic sites, as to occasionally make it difficult to identify individual malignant cells. In the signet ring variety, most of the tumor cells are distended by intracellular mucin, pushing the nucleus to the side. Tumor cells are more likely to infiltrate diffusely through the colonic wall, producing a linitis plastica of the affected colon. Signet ring mucinous colorectal carcinoma has a strong association with ulcerative colitis and HNPCC.
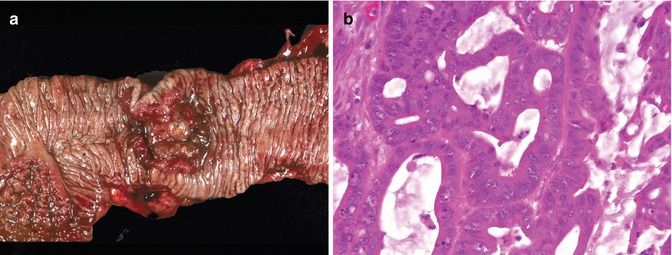
Fig. 8.14
Moderately differentiated colonic adenocarcinoma arising in a 15-year-old boy. (a) Ulcerated tumor with heaped-up borders. (b) Back-to-back malignant glands with prominent mucin production
8.3.5.6 Treatment
As in adults, complete surgical excision is the treatment of choice and offers the highest likelihood of cure. Unfortunately, in many cases the disease is advanced at diagnosis, precluding complete excision. As in adults, chemotherapy is also employed in primary therapy.
8.3.6 Neuroendocrine (Carcinoid) Tumors
8.3.6.1 Etiology
Carcinoid tumors are well-differentiated neuroendocrine epithelial tumors, not infrequently encountered in appendectomy specimens, but rarely seen in other parts of the gastrointestinal tract in children. In most instances, carcinoids are discovered in appendectomies performed clinically for acute appendicitis, and the finding of a carcinoid tumor is unexpected. Appendiceal carcinoids were found in 0.2–0.8 % of resected appendices in two pediatric series (Volpe et al. 2000; Doede et al. 2000). They are three times more frequent in girls than in boys (Spunt et al. 2000).
The rare carcinoid tumors that arise elsewhere in the alimentary tract may behave in a more aggressive fashion. Review of the surgical pathology records of the Children’s Hospital of Philadelphia uncovered a carcinoid tumor arising in a Meckel’s diverticulum, and carcinoids have also been described in rectal (Rubin et al. 1981) and gastric duplications (Horie et al. 1986). In a series of four malignant extra-appendiceal pediatric carcinoids, two arose in the gastrointestinal tract (ileum and transverse colon) and were both associated with extensive metastatic disease (Chow et al. 1982). A recent report describes a jejunal-based carcinoid tumor that presented in a 10-year-old boy with bilateral renal metastases. The patient subsequently developed liver and lung metastases and died at age 23 from intestinal obstruction (Warrier et al. 2011).
8.3.6.2 Clinical Manifestations
Appendiceal carcinoids may clinically mimic or coexist with acute appendicitis, especially when occurring proximal to the tip. Another common presentation is chronic abdominal pain. It seems likely that the majority of appendiceal carcinoids are clinically silent, with an age incidence in children not unexpectedly mirroring that of acute appendicitis. Nearly all appendiceal carcinoids in children are nonfunctioning (Coffin and Pappin 1997).
8.3.6.3 Pathology
Grossly, most carcinoids are small, less than 1 cm in size, yellow to tan on cut section, and may be nodular or diffusely thicken the appendiceal wall, sometimes circumferentially. They may grow into and sometimes obliterate the appendiceal lumen. The histologic appearance is typical of carcinoids found elsewhere in the body, with uniform, polygonal to slightly spindly tumor cells disposed in sheets, trabeculae, and nests (Fig. 8.15). Nuclei are round and uniform, chromatin is speckled, nucleoli are small, and mitoses are infrequent. Immunohistochemically they strongly express chromogranin and cytokeratin, and often will stain with other neuroendocrine markers. Ultrastructurally they typically contain pleomorphic neurosecretory granules. Local lymphatic, perineural, muscularis propria, and serosal invasion are all common features.
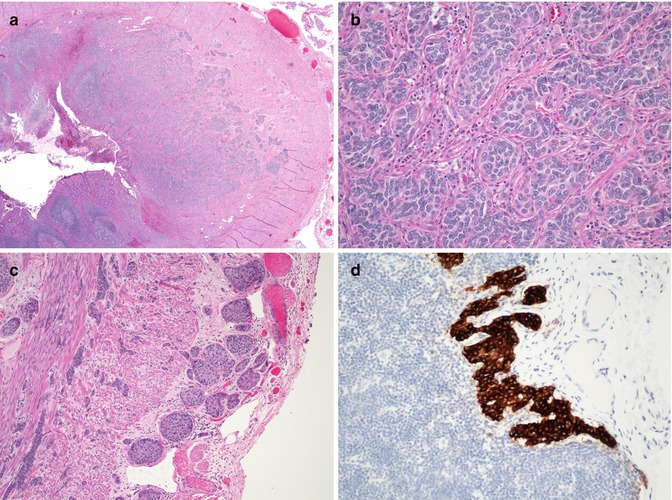
Fig. 8.15
Carcinoid tumor of appendix. (a) Low-power photomicrograph demonstrating an infiltrative neoplasm centered in the mucosa and submucosa. (b) Typical nesting pattern of monotonous polygonal neuroendocrine cells. (c) Photomicrograph of the same tumor, demonstrating invasion of the muscularis propria and serosa, a common finding in these tumors. (d) Immunohistochemical stain for chromogranin demonstrating a microscopic metastasis to a periappendiceal lymph node
8.3.6.4 Treatment
Although the histologic diagnosis of appendiceal carcinoid tumor is usually straightforward, differences of opinion exist as to the proper follow-up of such a diagnosis. In eight clinical series from disparate institutions in Europe and North America totaling 194 cases of pediatric appendiceal carcinoid tumors, there were no deaths attributed to the disease. Most patients did not have any surgery beyond their original appendectomies (Prommegger et al. 2002; Dall’Igna et al. 2005; Hatzipantelis et al. 2010) (Doede et al. 2000; Andersson and Bergdahl 1977; Corpron et al. 1995; Parkes et al. 1993; Ryden et al. 1975). Neither tumor size, depth of invasion or perineural involvement had prognostic significance (Andersson and Bergdahl 1977; Ryden et al. 1975). The majority of tumors were located at the tip of the appendix. In one study, fully 85 % of carcinoids had invaded beyond the appendiceal submucosa, which had no adverse prognostic implications (Parkes et al. 1993).
Recommendations in the literature, based on data largely accrued from adult patients, suggest segmental ileocolectomy for appendiceal carcinoids if they are greater than 2 cm in size, if they occur at the base, if they extend to the resection margin or the mesoappendix, if they display lymphatic invasion, or if they display mucinous histology (Moertel et al. 1987). Even in adults, these recommendations are controversial, as regional lymph node metastases are not uncommon even in appendiceal carcinoids measuring less than 2 cm in diameter (Mullen and Savarese 2011). The clinical significance of these regional nodal metastases is also unclear, as their presence seems to have little, if any, adverse effect on survival (Mullen and Savarese 2011).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


