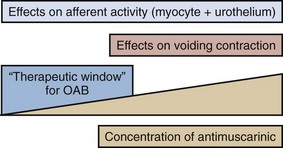
Karl-Erik Andersson, MD, PhD, Alan J. Wein, MD, PhD (Hon), FACS The function of the lower urinary tract (LUT) is to effect efficient and low-pressure bladder filling, low-pressure urine storage with normal sensation and perfect continence, and periodic complete voluntary emptying, again at low pressure. The structures involved include the smooth musculature of the bladder and the bladder outlet and the striated muscle, both intrinsic (to the bladder outlet) and extrinsic (the striated musculature surrounding the bladder outlet and the striated musculature of the pelvic floor). These component structures are controlled by a complex interplay between the central and peripheral nervous systems and local regulatory factors. Pharmacologic alteration of LUT function can occur at any point along the afferent or efferent limb of this complex neuromuscular cascade, either by receptor activation/stimulation or blockade, by affecting the concentration of neurotransmitter at an activation site, or by stimulating or inhibiting signal transduction mechanisms (Andersson and Wein, 2004; Andersson et al, 2009; Birder et al, 2009; Fry et al, 2009; see also Chapter 60). The focus of this chapter is the pharmacologic management of bladder filling/storage and bladder emptying/voiding dysfunction. The conceptual basis is that of the expanded functional classification seen in Table 61–5 and the division of therapies in the relatively simple-minded manner of those that facilitate urine storage/bladder filling and those that facilitate bladder emptying/voiding (see Tables 61–3 and 61–4). Although the principles expressed are generally applicable to all ages, specifics concerning usage in the elderly and pediatric age groups are considered in detail in Chapters 75, 126, and 127. Specific information regarding the pharmacologic management of LUT dysfunction secondary to obstruction by benign prostatic enlargement is considered in detail in Chapter 91, whereas drug therapy for the treatment of bladder and pelvic pain disorders is considered in detail in Chapters 11 and 12. The major portion of the neurohumoral stimulus for physiologic bladder contraction is acetylcholine-induced stimulation of postganglionic parasympathetic muscarinic cholinergic receptor sites in the bladder (detrusor smooth muscle and possibly other sites) (see Chapter 60). Atropine and atropine-like agents will depress normal bladder contractions and involuntary bladder contractions of any cause (Andersson, 1988; 1993; Andersson and Wein, 2004). In patients with involuntary contractions, the volume to the first involuntary contraction will generally be increased, the amplitude of the contraction decreased, and the total bladder capacity increased (Jensen, 1981). Previously, it has been stated (Wein, 1998) that bladder compliance in normal individuals and in those with neurogenic detrusor overactivity (NDO; Abrams et al, 2002), in whom the initial slope of the filling curve on cystometry is normal before the involuntary contraction, does not seem to be significantly altered by antimuscarinic agents and that the effect of pure antimuscarinics in those patients who exhibit only decreased compliance had not been well studied. Regarding the subject of bladder tone during filling, Andersson (1999a, 1999b, 2004; Andersson and Yoshida, 2003) has pointed out that, although it is widely accepted that there is normally no sacral parasympathetic outflow to the bladder during filling, antimuscarinic drugs increase, and anticholinesterase inhibitors decrease, bladder capacity. Because antimuscarinic drugs do seem to affect the sensation of urgency during filling, this suggests an ongoing acetylcholine-mediated stimulation of detrusor tone (see later). If this is correct, agents inhibiting acetylcholine release or activity would be expected to contribute to bladder relaxation or the maintenance of low bladder tone during filling with a consequent decrease in filling/storage symptomatology unrelated to the occurrence of an involuntary contraction. Outlet resistance, at least as reflected by urethral pressure measurements, does not seem to be clinically affected. Although the antimuscarinic agents usually produce significant clinical improvement in patients with involuntary contractions and associated symptoms, generally only partial inhibition results. In many animal models, atropine only partially antagonizes the response of the whole bladder to pelvic nerve stimulation and of bladder strips to field stimulation, although it does completely inhibit the response of bladder smooth muscle to exogenous cholinergic stimulation. This phenomenon, which is called atropine resistance, is secondary to release of a transmitter other than acetylcholine (see Andersson, 1993; Andersson et al, 1999c; Andersson and Wein, 2004; see also Chapter 60). Atropine resistance is the most common hypothesis invoked to explain the clinical difficulty in eradicating involuntary contractions with antimuscarinic agents alone, and it is also invoked to support the rationale of combined treatment of DO with agents that have different mechanisms of action (Andersson, 2006). Andersson and Wein (2004) cite references stating that atropine resistance seems to be of little importance in normal human bladder muscle but point out that atropine-resistant (nonadrenergic, noncholinergic) contractions have been reported in human detrusor smooth muscle and in morphologically and/or functionally changed bladders in individuals with various types of voiding dysfunction. Thus, the importance or nonimportance of an atropine-resistant component to detrusor contraction in the treatment of DO in humans remains to be established. In the human bladder, where the mRNAs for all the five pharmacologically defined receptors, M1 to M5, have been demonstrated (Sigala et al, 2002; Abrams et al, 2006a; Giglio and Tobin, 2009), there is a predominance of mRNAs encoding M2 and M3 receptors (Yamaguchi et al, 1996; Sigala et al, 2002; Abrams et al, 2006a; Giglio and Tobin, 2009). This seems to be the case also in the animal species investigated (Hegde and Eglen, 1999; Chess-Williams, 2002; Andersson and Arner, 2004). Both M2 and M3 receptors can be found on detrusor muscle cells, where M2 receptors predominate at least 3 : 1 over M3 receptors, but also in other bladder structures, which may be of importance for detrusor activation. Thus, muscarinic receptors can be found on urothelial cells, on suburothelial nerves, and on other suburothelial structures, such as interstitial cells (Chess-Williams, 2002; Gillespie et al, 2003; Gillespie, 2004b; Mansfield et al, 2005; Bschleipfer et al, 2007; Giglio and Tobin, 2009). In human as well as animal detrusor, the M3 receptors are believed to be the most important for contraction (Andersson, 1993; Chess-Williams, 2002; Abrams et al, 2006a; Giglio and Tobin, 2009). No differences between genders could be demonstrated in rat and human bladders (Kories et al, 2003). The functional role for the M2 receptors has not been clarified, and even in M3 receptor knockout mice they seem responsible for less that 5% of the carbachol-mediated detrusor contraction (Matsui et al, 2000). Stimulation of M2 receptors has been shown to oppose sympathetically (β-adrenoreceptor [β-AR]) mediated smooth muscle relaxation (Hegde et al, 1997). However, based on animal experiments, M2 receptors have been suggested to directly contribute to contraction of the bladder in certain disease states (denervation, outflow obstruction). Experiments on human detrusor muscle by Stevens and associates (2007) could not confirm this; however, Pontari and colleagues (2004) analyzed bladder muscle specimens from patients with neurogenic bladder dysfunction to determine whether the muscarinic receptor subtype mediating contraction shifts from M3 to the M2 receptor subtype, as found in the denervated, hypertrophied rat bladder. They concluded that normal detrusor contraction is mediated by the M3 receptor subtype whereas contractions can be mediated by the M2 receptors in patients with neurogenic bladder dysfunction. Muscarinic receptors are coupled to G proteins, but the signal transduction systems may vary. Generally, M1, M3, and M5 receptors are considered to couple preferentially to Gq/11, activating phosphoinositide hydrolysis, in turn leading to mobilization of intracellular calcium (Ca2+). M2 and M4 receptors couple to pertussis toxin–sensitive Gi/o, resulting in inhibition of adenylate cyclase activity. In the human detrusor, Schneider and coworkers (2004), confirming that the muscarinic receptor subtype mediating carbachol-induced contraction is the M3 receptor, also demonstrated that the phospholipase-C inhibitor U73122 did not significantly affect carbachol-stimulated bladder contraction, despite blocking inositol 1,4,5-triphosphate generation. They concluded that carbachol-induced contraction of the human urinary bladder is mediated via M3 receptors and largely depends on Ca2+ entry through nifedipine-sensitive channels and activation of the Rho-kinase pathway. Thus, it may be that the main pathways for muscarinic receptor activation of the detrusor via M3 receptors are Ca2+ influx via L-type Ca2+ channels and increased sensitivity to Ca2+ of the contractile machinery via inhibition of myosin light-chain phosphatase through activation of Rho-kinase. The signaling mechanisms for the M2 receptors are less clear than those for M3 receptors. As mentioned previously, M2 receptor stimulation may oppose sympathetically induced smooth muscle relaxation, mediated by β-ARs via inhibition of adenylyl cyclase (Hegde et al, 1997). In agreement with this, Matsui and colleagues (2003) suggested, based on results obtained in M2 receptor knockout mice, that a component of the contractile response to muscarinic agonists in smooth muscle involves an M2 receptor–mediated inhibition of the relaxant effects of agents that increase cyclic adenosine monophosphate (cAMP) levels. M2 receptor stimulation can also activate nonspecific cation channels and inhibit KATP channels through activation of protein kinase C (Bonev and Nelson, 1993; Kotlikoff et al, 1999). Muscarinic receptors may also be located on the presynaptic nerve terminals and participate in the regulation of transmitter release. The inhibitory prejunctional muscarinic receptors have been classified as muscarinic M2 in the rabbit (Tobin and Sjögren, 1995) and rat (Somogyi and de Groat, 1992) and as M4 in the guinea pig (Alberts, 1995) and human bladder (D’Agostino et al, 2000). Prejunctional facilitatory muscarinic receptors appear to be of the M1 subtype in the bladders of rats, rabbits (Somogyi and de Groat, 1992; Tobin and Sjögren, 1995, 1998), and humans (Somogyi and de Groat, 1999; Giglio and Tobin, 2009). The muscarinic facilitatory mechanism seems to be upregulated in overactive bladders from chronic spinal cord–transected rats. The facilitation in these preparations is primarily mediated by M3 muscarinic receptors (Somogyi and de Groat, 1999). The relative roles of the different presynaptic and postsynaptic receptor subtypes in normal and abnormal bladder function still require clarification, and thus speculation regarding optimal drug therapy based only on in-vitro receptor selectivity profiles represents, at the very least, a gross oversimplification of assumptions regarding the muscarinic regulation of bladder function. The muscarinic receptor functions may be changed in different urologic disorders, such as outflow obstruction, neurogenic bladders, DO without overt neurogenic cause, and diabetes (Andersson, 2000b). However, it is not always clear what the changes mean in terms of changes in detrusor function. In general, all drug therapy for LUT dysfunction is hindered by a concept that can be expressed in one word: “uroselectivity” (Andersson, 1998). The clinical utility of available antimuscarinic agents is limited by their lack of selectivity, which is responsible for the classic peripheral antimuscarinic side effects of dry mouth, constipation, blurred vision, and increase in heart rate and for the effects on cognitive functions. Although M3 receptor selective agents have the potential to eliminate some of these side effects, it would appear that the M3 receptors in tissues of the LUT are identical to those elsewhere in the body (Caulfield and Birdsall, 1998). It may be speculated, however, that there is some heterogeneity among M3 receptors, and this has prompted many pharmaceutical companies to continue to search for the “ideal” antimuscarinic agent to treat DO, one that would be relatively selective for muscarinic receptors involved in the regulation of bladder contraction. Receptor selectivity, however, is not the only basis on which a drug may be “uroselective.” From a clinical standpoint it would seem particularly important to be able to describe in relative terms the ratio between a drug dose required for a desired therapeutic action and the dose producing side effects. A differential effect could be based not only on receptor selectivity but also on other known and as yet undefined physiologic, pharmacologic, or metabolic characteristics. Organ selectivity would thus seem to be the “holy grail” of such therapy. The same problematic set of concepts applies to virtually all drugs used for the treatment of LUT dysfunction. The prevalence of the overactive bladder (OAB) syndrome varies with the criteria for diagnosis. According to Irwin and associates (2006), using the 2002 International Continence Society (ICS) definition, the overall prevalence (the EPIC study) was 11.8%. The rates were similar in men and women and increased with age. A similar study by Herschhorn and coworkers (2008) found the prevalence to be 13% in Canadian men and 14% in Canadian women. It appears that OAB/DO may be the result of several different mechanisms, both myogenic and neurologic (Morrison et al, 2002; Birder et al, 2009; Koebl et al, 2009). Most probably, both factors contribute to the genesis of the disease. An abundance of drugs has been used for the treatment of OAB/DO. It should be stressed that in many trials on OAB/DO there has been such a high placebo response that meaningful differences between placebo and active drug cannot be demonstrated (Thüroff et al, 1998). However, drug effects in individual patients may be both distinct and useful. The 4th International Consultation on Incontinence (ICI) (2008) assessed drugs used for treatment of incontinence (Andersson et al, 2009). The assessment criteria (Table 68–1) were based on the Oxford guidelines, and the drugs included are given in Table 68–2. Table 68–1 International Consultation on Incontinence (ICI) Assessments 2004, 2008: Oxford Guidelines (Modified) Table 68–2 Drugs Used in the Treatment of Detrusor Overactivity (ICI, 2008; Andersson et al, 2009) Acetylcholine stimulates both muscarinic and nicotinic receptors. Antimuscarinic agents block selectively muscarinic receptors, and they are currently the mainstay of treatment of OAB/DO (Abrams and Andersson, 2007). The traditional view is that in OAB/DO the drugs act by blocking the muscarinic receptors on the detrusor muscle, which are stimulated by acetylcholine released from activated cholinergic (parasympathetic) nerves. Thereby, they decrease the ability of the bladder to contract. However, antimuscarinic drugs act mainly during the storage phase, decreasing urgency and increasing bladder capacity; and during this phase there is normally no parasympathetic input to the LUT (Morrison et al, 2002; Andersson, 2004). Furthermore, antimuscarinic agents are usually competitive antagonists. This implies that when there is a massive release of acetylcholine, as during micturition, the effects of the drugs should be decreased, otherwise the reduced ability of the detrusor to contract would eventually lead to urinary retention. Undeniably, high doses of antimuscarinic agents can produce urinary retention in humans, but in the dose range used for beneficial effects in OAB/DO there is little evidence for a significant reduction of the voiding contraction (Finney et al, 2006) (Fig. 68–1). There is indirect clinical evidence for release of acetylcholine during bladder filling in certain abnormal conditions. Smith and colleagues (1974) found that in patients with recent spinal cord injury, inhibition of acetylcholine breakdown by use of cholinesterase inhibitors could increase resting tone and induce rhythmic contractions in the bladder. Yossepowitch and coworkers (2001) inhibited acetylcholine breakdown with edrophonium in a series of patients with disturbed voiding or urinary incontinence. They found a significant change in sensation and decreased bladder capacity, induction or amplification of involuntary detrusor contractions, or significantly decreased detrusor compliance in 78% of the patients with the symptom pattern of OAB but in no patients without specific complaints suggesting DO. Thus, during the storage phase, acetylcholine may be released from both neuronal and non-neuronal sources (e.g., the urothelium/suburothelium) and directly or indirectly (by increasing detrusor smooth muscle tone) excite afferent nerves in the suburothelium and within the detrusor. There is also good experimental evidence that antimuscarinic agents act during the storage phase by decreasing the activity in afferent nerves (both C and Aδ fibers) from the bladder (De Laet et al, 2006; Iijima et al, 2007). Muscarinic receptors are found on bladder urothelial cells where their density can be even higher than in detrusor muscle. The role of the urothelium in bladder activation has attracted much interest (Andersson, 2002a; de Groat, 2004; Birder and de Groat, 2007; Birder et al, 2009; Giglio and Tobin, 2009), but whether the muscarinic receptors on urothelial cells can influence micturition has not yet been established. Yoshida and colleagues (2004, 2006, 2008) found that there is basal acetylcholine release in the human bladder. This release was resistant to tetrodotoxin and much diminished when the urothelium was removed; thus, the released acetylcholine was probably of non-neuronal origin and, at least partly, generated by the urothelium. Thus, during the storage phase, acetylcholine and adenosine triphosphate (ATP) may be released from both neuronal and non-neuronal sources (e.g., the urothelium) and directly or indirectly (by increasing detrusor smooth muscle tone) excite afferent nerves in the suburothelium and within the detrusor. These mechanisms may be important in the pathophysiology of OAB and represent possible targets for antimuscarinic drugs. Generally, antimuscarinic agents can be divided into tertiary and quaternary amines (Guay, 2003; Abrams and Andersson, 2007). They differ with regard to lipophilicity, molecular charge, and even molecular size, with tertiary compounds generally having higher lipophilicity and molecular charge than quaternary agents. Atropine, darifenacin, fesoterodine (and its active metabolite 5-hydroxymethyl-tolterodine), oxybutynin, propiverine, solifenacin, and tolterodine, are tertiary amines. They are generally well absorbed from the gastrointestinal tract and should theoretically be able to pass into the central nervous system (CNS), dependent on their individual physicochemical properties. High lipophilicity, small molecular size, and less charge will increase the possibilities to pass the blood-brain barrier, but for some of the drugs this is counteracted by active transport out of the CNS by P-glycoprotein. Quaternary ammonium compounds, such as propantheline and trospium, are not well absorbed, pass into the CNS to a limited extent, and have a low incidence of CNS side effects (Guay, 2003). They still produce well-known peripheral antimuscarinic side effects, such as accommodation paralysis, constipation, increase in heart rate, and dryness of mouth. Many antimuscarinic drugs are metabolized by the cytochrome P450 enzyme system to active and/or inactive metabolites (Guay, 2003). The most commonly involved P450 enzymes are CYP2D6 and CYP3A4. The metabolic conversion creates a risk for drug-drug interactions, resulting in either reduced (enzyme induction) or increased (enzyme inhibition, substrate competition) plasma concentration/effect of the antimuscarinic and/or interacting drug. Antimuscarinic agents secreted by the renal tubules (e.g., trospium) may theoretically be able to interfere with the elimination of other drugs using this mechanism. Antimuscarinic drugs are still the most widely used treatment for urgency and urgency incontinence (Andersson, 2004; Andersson et al, 2009). However, currently used drugs lack selectivity for the bladder, and effects on other organ systems (Table 68–3) may result in side effects that limit their usefulness. For example, all antimuscarinic drugs are contraindicated in patients with untreated narrow-angle glaucoma. Table 68–3 Muscarinic Receptors: Distribution and Function Data from Abrams P, Andersson K-E, Buccafusco JJ, et al. Muscarinic receptors: their distribution and function in body systems, and the implications for treating overactive bladder. Br J Pharmacol 2006;148:565. Several antimuscarinic drugs are and have been used for treatment of OAB/DO. For many of them documentation of effects is not based on randomized controlled trials (RCTs) satisfying currently required criteria, and some drugs can be considered as obsolete (e.g., emepronium). Information on these drugs is found elsewhere (Andersson, 1988; Andersson et al, 1999c). The clinical relevance of efficacy of antimuscarinic drugs relative to placebo has been questioned. Herbison and associates (2003) stated in a widely discussed article: “Anticholinergics produce significant improvements in overactive bladder symptoms compared with placebo. The benefits are, however, of limited clinical significance.” Large meta-analyses of studies performed with the currently most widely used drugs (Chapple et al, 2005, 2008a; Novara et al, 2008) clearly show that antimuscarinic drugs are of significant clinical benefit. Novara and colleagues (2008) reviewed 50 RCTs and 3 pooled analyses that they considered of good methodologic quality. They concluded that still more clinical studies are needed to decide which of the drugs should be used as first-, second-, or third-line treatment. Reviewing information from more than 12,000 references, Chapple and colleagues (2008a) based their conclusions (“antimuscarinics are efficacious, safe, and well tolerated treatments”) on 73 RCTs selected for their meta-analysis. It was recommended that because the profiles of each drug and dosage differ that these factors should be considered in making treatment choices. The consequence of this is that, in the authors’ opinion, none of the antimuscarinic drugs in common clinical use (darifenacin, fesoterodine, oxybutynin, propiverine, solifenacin, tolterodine, or trospium) is ideal as a first-line treatment for all OAB/DO patients. Optimal treatment should be individualized, implying that the patient’s comorbidities and concomitant medications, and the pharmacologic profiles of the different drugs, should be taken into consideration (Chapple et al, 2008a). Behavioral therapy (see Chapter 69) should always be used in conjunction with drug therapy for OAB/DO because most studies show the effects of the two combined are greater than the effect of either alone (Burgio et al, 2000; Mattiasson et al, 2003; Klutke et al, 2009). Among the more serious possibilities related to antimuscarinic use is the potential risk of cardiac adverse effects, particularly increases in heart rate and QT prolongation and induction of polymorphic ventricular tachycardia (torsades de pointes). QT prolongation and its consequences are not related to blockade of muscarinic receptors but rather linked to inhibition of the hERG potassium (K+) channel in the heart (Roden, 2004). Thus QT prolongation is not a class effect of antimuscarinic drugs. In general the cardiovascular safety for antimuscarinic drugs seems to be good. However, the potential of the different agents to increase heart rate or to prolong the QT time has not been extensively explored. Differences between the drugs cannot be excluded, but risk assessments based on available evidence are not possible. Atropine (dl-hyoscyamine) is rarely used for treatment of OAB/DO because of its systemic side effects, which preclude its use as an oral treatment. However, in patients with neurogenic DO, intravesical atropine may be effective for increasing bladder capacity without causing any systemic adverse effects, as shown in open pilot trials (Ekström et al, 1992; Glickman et al, 1995; Deaney et al, 1998; Enskat et al, 2001; Fader et al, 2007). It appears that intravesical atropine may be as effective as intravesical oxybutynin in patients with neurogenic DO (Fader et al, 2007). The pharmacologically active antimuscarinic component of atropine is l-hyoscyamine. Although it is still used, few clinical studies are available to evaluate the antimuscarinic activity of l-hyoscyamine sulfate (Muskat et al, 1996). For assessment, see Table 68–2. Propantheline bromide (Pro-Banthine, others) is a quaternary ammonium compound, nonselective for muscarinic receptor subtypes, that has a low (5% to 10%) and individually varying biologic availability. It is metabolized (metabolites inactive) and has a short plasma half-life (<2 hr) (Beermann et al, 1972). It is usually given in a dose of 15 to 30 mg four times daily, but, to obtain an optimal effect, individual titration of the dose is necessary and often higher dosages are required. Using this approach in 26 patients with DO, Blaivas and associates (1980) in an open study obtained a complete clinical response in all patients but one, who did not tolerate more than propantheline, 15 mg, four times daily. The range of dosages varied from 7.5 to 60 mg four times daily. In contrast, Thüroff and coworkers (1991) comparing the effects or oxybutynin 5 mg three times a day, propantheline 15 mg three times a day, and placebo in a randomized, double-blind, multicenter trial on the treatment of frequency, urgency, and incontinence related to DO (154 patients) found no differences between the placebo and propantheline groups. In another randomized comparative trial with crossover design (23 women with idiopathic DO), and with dose titration, Holmes and associates (1989) found no differences in efficacy between oxybutynin and propantheline. There is a surprising lack of evaluable data on the effectiveness of propantheline for the treatment of DO. The Agency for Health Care Policy and Research (AHCPR) Clinical Practice Guidelines (U.S. Department of Health and Human Services, 1992) lists five randomized controlled trials reviewed for propantheline, with 82% female patients. Percent cures (all figures refer to percent effect on drug minus percent effect on placebo) are listed as 0% to 5%, reduction in urgency incontinence as 0% to 53%, and percent side effects and percent dropouts as 0% to 50% and 0% to 9%, respectively. RCTs (n = 6) reviewed by Thüroff and associates (1998) confirmed a positive, but varying, response to the drug. Although the effect of propantheline on OAB/DO has not been well documented in controlled trials satisfying standards of today, it can be considered effective and may, in individually titrated doses, be clinically useful (see Table 68–2). No new studies on the use of this drug for the treatment of OAB/DO seem to have been performed during the past decade. Trospium is a quaternary ammonium compound with a biologic availability of less than 10% (Fusgen and Hauri, 2000; Doroshyenko et al, 2005). The drug has a plasma half-life of approximately 20 hours and is mainly (60% of the dose absorbed) eliminated unchanged in the urine. The concentration obtained in urine seems to be enough to affect the mucosal signaling system in a rat model (Kim et al, 2006). Whether it contributes to the clinical efficacy of the drug remains to be established. Trospium is not metabolized by the cytochrome P450 enzyme system (Beckmann-Knopp et al, 1999; Doroshyenko et al, 2005). It is expected to cross the blood-brain to a limited extent and seems to have no negative cognitive effects (Fusgen and Hauri, 2000; Todorova et al, 2001; Wiedemann et al, 2002). Trospium has no selectivity for muscarinic receptor subtypes. In isolated detrusor muscle it was more potent than oxybutynin and tolterodine to antagonize carbachol-induced contractions (Uckert et al, 2000). Several RCTs have documented positive effects of trospium both in neurogenic (Stöhrer, et al, 1991; Madersbacher et al, 1995; Menarini et al, 2006) and non-neurogenic DO (Allousi et al, 1998; Cardozo et al, 2000; Jünemann and Al-Shukri, 2000; Halaska et al, 2003; Zinner et al, 2004a; Rudy et al, 2006; Staskin et al, 2007; Dmochowski et al, 2008). In a placebo-controlled, double-blind study on patients with neurogenic DO (Stöhrer et al, 1991) the drug was given twice daily in a dose of 20 mg over a 3-week period. It increased maximum cystometric capacity, decreased maximum detrusor pressure, and increased compliance in the treatment group, whereas no effects were noted in the placebo group. Side effects were few and comparable in both groups. In another RCT including patients with spinal cord injuries and neurogenic DO, trospium and oxybutynin were equally effective; however, trospium seemed to have fewer side effects (Madersbacher et al, 1995). The effect of trospium in urgency incontinence has been documented in several RCTs. Allousi and associates (1998) compared the effects of the drug with those of placebo in 309 patients in a urodynamic study of 3 weeks’ duration. Trospium, 20 mg, was given twice daily. Significant increases were noted in volume at first involuntary contraction and in maximum bladder capacity. Cardozo and coworkers (2000) investigated 208 patients with DO who were treated with trospium, 20 mg twice daily, for 2 weeks. Also in this study, significant increases were found in mean volume at first unstable contraction (from 233 to 299 mL; placebo 254 to 255 mL) and in maximum bladder capacity (from 329 to 356 mL; placebo 345 to 335 mL) in the trospium-treated group. Trospium was well tolerated with similar frequency of adverse effects as in the placebo group. Jünemann and Al-Shukri (2000) compared trospium, 20 mg twice daily, with tolterodine, 2 mg twice daily, in a placebo-controlled double-blind study on 232 patients with urodynamically proven DO, urgency incontinence without demonstrable DO, or mixed incontinence. Trospium reduced the frequency of micturition, which was the primary end point, more than tolterodine and placebo and also reduced the number of incontinence episodes more than the comparators. Dry mouth was comparable in the trospium and tolterodine groups (7% and 9%, respectively). Halaska and colleagues (2003) studied the tolerability and efficacy of trospium chloride in doses of 20 mg twice daily for long-term therapy in patients with urgency syndrome. The trial comprised a total of 358 patients with urgency syndrome or urgency incontinence. After randomization in the ratio of 3 : 1, participants were treated continuously for 52 weeks with either trospium chloride (20 mg twice daily) or oxybutynin (5 mg twice daily). Urodynamic measurements were performed at the beginning and at 26 and 52 weeks to determine the maximum cystometric bladder capacity. Analysis of the micturition diary clearly indicated a reduction of the micturition frequency, incontinence frequency, and a reduction of the number of urgency episodes in both treatment groups. Mean maximum cystometric bladder capacity increased during treatment with trospium chloride by 92 mL after 26 weeks and 115 mL after 52 weeks (P = .001). Further comparison with oxybutynin did not reveal any statistically significant differences in urodynamic variables between the drugs. Adverse events occurred in 65% of the patients treated with trospium and 77% of those treated with oxybutynin. The main symptom encountered in both treatment groups was dryness of the mouth. An overall assessment for each of the drugs revealed a comparable efficacy level and a better benefit-risk ratio for trospium than for oxybutynin due to better tolerability. Zinner and coworkers (2004a) treated 523 patients with symptoms associated with OAB and urgency incontinence with trospium 20 mg twice daily or placebo in a 12-week, multicenter, parallel, double-blind, placebo-controlled trial. Dual primary end points were change in average number of toilet voids and change in urgency incontinent episodes per 24 hours. Secondary efficacy variables were change in average of volume per void, voiding urgency severity, urinations during day and night, time to onset of action, and change in Incontinence Impact Questionnaire. By week 12, trospium significantly decreased average frequency of toilet voids per 24 hours (−2.37; placebo −1.29) and urgency incontinent episodes (−59%; placebo −44%). It significantly increased average volume per void (32 mL; placebo: 7.7 mL) and decreased average urgency severity and daytime frequency. All effects occurred by week 1, and all were sustained throughout the study. Nocturnal frequency decreased significantly by week 4 (−0.43; placebo: 0.17), and Incontinence Impact Questionnaire scores improved at week 12. Trospium was well tolerated. The most common side effects were dry mouth (21.8%; placebo 6.5%), constipation (9.5%; placebo 3.8%). and headache (6.5%; placebo 4.6%). In a large U.S. multicenter trial with the same design, and including 658 patients with OAB, Rudy and coworkers (2006) confirmed the data by Zinner and associates (2004a), both with respect to efficacy and adverse effects. An extended-release formulation of trospium allowing once-daily dosing (60 mg) has been introduced and its effects tested in controlled trials (Staskin et al, 2007; Dmochowski et al, 2008). These RCTs demonstrated similar efficacy as found with previous formulations. The most frequent side effects were dry mouth (12.9%; placebo 4.6%) and constipation (7.5%; placebo 1.8%) (Dmochowski et al, 2008). Intravesical application of trospium may be an interesting alternative. Fröhlich and colleagues (1998) performed a randomized, single-blind, placebo-controlled, monocenter clinical trial in 84 patients with urgency or urgency incontinence. Compared with placebo, intravesical trospium produced a significant increase in maximum bladder capacity and a decrease of detrusor pressure accompanied by an increase of residual urine. There was an improvement in uninhibited bladder contractions. No adverse events were reported. Interestingly, intravesical trospium does not seem to be absorbed (Walter et al, 1999), thus offering an opportunity for treatment with minimal systemic antimuscarinic effects. Trospium is a well-documented alternative for treatment of OAB/DO and seems to be well tolerated. Tolterodine is a tertiary amine, rapidly absorbed and extensively metabolized by the cytochrome P450 system (CYP2D6). The major active 5-hydroxymethyl metabolite (5-HMT) has a similar pharmacologic profile as the mother compound (Nilvebrant et al, 1997b) and significantly contributes to the therapeutic effect of tolterodine (Brynne et al, 1997, 1998). Both tolterodine and 5-HMT have plasma half-lives of 2 to 3 hours, but the effects on the bladder seem to be more long lasting than could be expected from the pharmacokinetic data. Urinary excretion of tolterodine accounted for less than 1.0% to 2.4% of the dose; 5% to 14% of 5-HMT is eliminated in the urine (Brynne et al, 1997). Whether the total antimuscarinic activity of unchanged tolterodine and 5-HMT excreted in urine is sufficient to exert any effect on the mucosal signaling mechanisms has not been established. However, the preliminary studies by Kim and coworkers (2006) and Chuang and colleagues (2008) do not support such an effect. The relatively low lipophilicity of tolterodine and even lesser one of 5-HMT implies limited propensity to penetrate into the CNS, which may explain a low incidence of cognitive side effects (Hills et al, 1998; Clemett et al, 2001; Salvatore et al, 2008). However, tolterodine may disturb sleep in subjects unable to form the even less lipophilic 5-HMT due to a low activity of CYP2D6 (Diefenbach et al, 2008). Tolterodine has no selectivity for muscarinic receptor subtypes but is claimed to have functional selectivity for the bladder over the salivary glands (Stahl et al, 1995; Nilvebrant et al, 1996, 1997a, 1997c). In healthy volunteers, orally given tolterodine in a high dose (6.4 mg) had a powerful inhibitory effect on micturition and also reduced stimulated salivation 1 hour after administration of the drug (Stahl et al, 1995); however, 5 hours after administration, the effects on the urinary bladder were maintained whereas no significant effects on salivation could be demonstrated. Tolterodine is available as immediate-release (TOLT-IR; 1 or 2 mg; twice-daily dosing) and extended-release (TOLT-ER) forms (2 or 4 mg; once-daily dosing). The ER form seems to have advantages over the IR form in terms of both efficacy and tolerability (Van Kerrebroeck et al, 2001). Several randomized, double-blind, placebo-controlled studies on patients with OAB/DO (both idiopathic and neurogenic DO) have documented a significant reduction in micturition frequency and number of incontinence episodes (Hills et al, 1998; Clemett et al, 2001; Salvatore et al, 2008). Comparative RCTs such as the OBJECT (Overactive Bladder: Judging Effective Control and Treatment) and the OPERA (Overactive Bladder; Performance of Extended Release Agents) studies have further supported its effectiveness. The OBJECT trial compared oxybutynin ER (OXY-ER) 10 mg once daily with TOLT-IR 2 mg twice daily (Appell et al, 2001) in a 12-week randomized, double-blind, parallel-group study including 378 patients with OAB. Participants had between 7 and 50 episodes of urgency incontinence per week and 10 or more voids in 24 hours. The outcome measures were the number of episodes of urgency incontinence, total incontinence, and micturition frequency at 12 weeks adjusted for baseline. At the end of the study, OXY-ER was found to be significantly more effective than TOLT-IR in each of the main outcome measures adjusted for baseline (see also later discussion on oxybutynin chloride). Dry mouth, the most common adverse event, was reported by 28% and 33% of participants taking OXY-ER and TOLT-IR, respectively. Rates of CNS and other adverse events were low and similar in both groups. The authors concluded that OXY-ER was more effective than TOLT-IR and that the rates of dry mouth and other adverse events were similar in both treatment groups. In the OPERA study (Diokno et al, 2003), OXY-ER at 10 mg/day or TOLT-ER at 4 mg/day was given for 12 weeks to women with 21 to 60 urgency incontinence episodes per week and an average of 10 or more voids per 24 hours. Episodes of incontinence episodes (primary end point), total (urgency and nonurgency) incontinence, and micturition were recorded in seven 24-hour urinary diaries at baseline and at weeks 2, 4, 8, and 12 and compared. Adverse events were also evaluated. Improvements in weekly urgency incontinence episodes were similar for the 790 women who received OXY-ER (n = 391) or TOLT-ER (n = 399). OXY-ER was significantly more effective than TOLT-ER in reducing micturition frequency, and 23.0% of women taking OXY-ER reported no episodes of urinary incontinence compared with 16.8% of women taking TOLT-ER. Dry mouth (usually mild) was more common with OXY-ER. Adverse events were generally mild and occurred at low rates, with both groups having similar discontinuation of treatment due to adverse events. The conclusions were that reductions in weekly urgency incontinence and total incontinence episodes were similar with the two drugs. Dry mouth was more common with OXY-ER, but tolerability was otherwise comparable, including adverse events involving the CNS. In the ACET (Antimuscarinic Clinical Effectiveness Trial) (Sussman and Garely, 2002) study, which consisted of two trials, patients with OAB were randomized to 8 weeks of open-label treatment with either 2 mg or 4 mg of once-daily TOLT-ER (study 1) and to 5 mg or 10 mg of OXY-ER (study 2). A total of 1289 patients were included. Fewer patients prematurely withdrew from the trial in the TOLT-ER 4-mg group (12%) than either the OXY-ER 5-mg (19%) or OXY-ER 10-mg groups (21%). More patients in the OXY-ER 10-mg group than the TOLT-ER 4-mg group withdrew because of poor tolerability (13% vs. 6%). After 8 weeks, 70% of patients in the TOLT-ER 4-mg group perceived an improved bladder condition, compared with 60% in the TOLT-ER 2-mg group, 59% in the OXY-ER 5-mg group and 60% in the OXY-ER 10-mg group. Dry mouth was dose dependent with both agents, although differences between doses reached statistical significance only in the oxybutynin trial (OXY-ER 5 mg vs. OXY-ER 10 mg; P = .05). Patients treated with TOLT-ER 4 mg reported a significantly lower severity of dry mouth compared with OXY-ER 10 mg. The conclusion that the findings suggest improved clinical efficacy of TOLT-ER (4 mg) over OXY-ER (10 mg) is weakened by the open-label design of the study. Zinner and associates (2002) evaluated the efficacy, safety, and tolerability of TOLT-ER in older (≥65) and younger (<65) OAB patients in a 12-week RCT including 1015 patients with urgency incontinence and urinary frequency. Patients were randomized to treatment with TOLT-ER 4 mg once daily (n = 507) or placebo (n = 508) for 12 weeks. Efficacy, measured with micturition charts (incontinence episodes, micturitions, volume voided per micturition) and subjective patient assessments, safety, and tolerability end points were evaluated, relative to placebo. Compared with placebo, significant improvements in micturition chart variables with TOLT-ER showed no age-related differences. Dry mouth (of any severity) was the most common adverse event in both the TOLT-ER and placebo treatment study arms, irrespective of age (<65: TOLT-ER 22.7%, placebo 8.1%; ≥65: TOLT-ER 24.3%, placebo 7.2%). A few patients (<2%) experienced severe dry mouth. No CNS (cognitive functions were not specifically studied), visual, cardiac (per electrocardiogram), or laboratory safety concerns were noted in this study. Withdrawal rates due to adverse events on TOLT-ER 4 mg once daily were comparable in the two age cohorts (<65: 5.5%; ≥65: 5.1%). The central symptom in the OAB syndrome is urgency. Freeman and coworkers (2003) presented a secondary analysis of a double-blind, placebo-controlled study evaluating the effect of once-daily TOLT-ER on urinary urgency in patients with OAB. Patients with urinary frequency (eight or more micturitions per 24 hours) and urgency incontinence (five or more episodes per week) were randomized to oral treatment with TOLT-ER 4 mg once daily (n = 398) or placebo (n = 374) for 12 weeks. Efficacy was assessed by use of patient perception evaluations. Of patients treated with TOLT-ER, 44% reported improved urgency symptoms (compared with 32% for placebo) and 62% reported improved bladder symptoms (placebo, 48%). The proportion of patients unable to hold urine on experiencing urgency was decreased by 58% with TOLT-ER, compared with 32% with placebo (P < .001). In the IMprovement in Patients: Assessing symptomatic Control with Tolterodine ER (IMPACT) study (Elinoff et al, 2006), the efficacy of TOLT-ER for patients’ most bothersome OAB symptom was investigated in an open-label, primary care setting. Patients with OAB symptoms for more than 3 months received TOLT-ER (4 mg once daily) for 12 weeks. By week 12 there were significant reductions in the patients’ most bothersome symptom—incontinence—urgency episodes, and nocturnal and daytime frequency. The most common adverse events were dry mouth (10%) and constipation (4%), and it was concluded that, in primary care practice, bothersome OAB symptoms can be effectively and safely treated with TOLT-ER, even in patients with comorbid conditions. Various aspects of the efficacy and tolerability of tolterodine have been further documented in a number of RCTs (Dmochowski et al, 2007a, 2007b; Barucha et al, 2008; Choo et al, 2008; Coyne et al, 2008; Rogers et al, 2008; Rovner et al, 2008b; for further discussion see Chapple et al, 2008b; Novara et al, 2008). Importantly, the QTc effects of tolterodine were determined in a crossover-designed QT study of recommended (2 mg twice daily) and supratherapeutic (4 mg twice daily) doses of tolterodine, moxifloxacin (400 mg once daily), and placebo. No subject receiving tolterodine exceeded the clinically relevant thresholds of 500 msec absolute QTc or 60 msec change from baseline, and it was concluded that tolterodine does not have a clinically significant effect on QT interval (Malhotra et al, 2007). Olshansky and colleagues (2008) compared, in a randomized, placebo-controlled, double blind crossover study, the effects on heart rate of TOLT-ER 4 mg/day with those of darifenacin 15 mg/day and placebo in 162 healthy volunteers. They found that tolterodine, but not darifenacin and placebo, significantly increased mean heart rate per 24 hours. The treatment difference in adjusted mean change from baseline was +1.84 beats per minute (P = .0004) for tolterodine versus darifenacin and +1.42 beats per minute (P < .001) for tolterodine versus placebo. The proportion of subjects with an increase greater than 5 beats per minute was significantly greater in those receiving TOLT-ER (1 of 4) than with darifenacin (1 of 10). The clinical significance of this has yet to be established. In a prospective, open study, Song and associates (2006) compared the effects of bladder training and/or tolterodine as first-line treatment in female patients with OAB. One hundred and thirty-nine female patients with OAB were randomized to treatment with bladder training, tolterodine (2 mg twice daily), or both for 12 weeks. All treatments were efficacious; however, combination therapy was the most effective. Mattiasson and colleagues (2003) compared the efficacy of tolterodine 2 mg twice daily plus simplified bladder training with tolterodine alone in patients with OAB in a multicenter single-blind study. At the end of the study the median percentage reduction in voiding frequency was greater with tolterodine plus bladder training than with tolterodine alone (33% vs. 25%; P < .001), whereas the median percentage increase in volume voided per void was 31% with tolterodine plus bladder training and 20% with tolterodine alone (P < .001). There was a median of 81% fewer incontinence episodes than at baseline with tolterodine alone, which was not significantly different from that with tolterodine plus bladder training (−87%). It was concluded that the effectiveness of tolterodine 2 mg twice daily can be augmented by a simplified bladder training regimen. However, Millard and colleagues (2004) investigated whether the combination of tolterodine plus a simple pelvic floor muscle exercise program would provide improved treatment benefits compared with tolterodine alone in 480 patients with OAB. Tolterodine therapy for 24 weeks resulted in significant improvement in urgency, frequency, and incontinence; however, no additional benefit was demonstrated for a simple pelvic floor muscle exercise program. The beneficial effect of TOLT-ER in men with benign prostatic enlargement and LUTS, including OAB, has been well documented. Both as monotherapy, but particularly in combination with an α-adrenergic receptor (AR) antagonist, TOLT-ER was found effective (Kaplan et al, 2006; Höfner et al, 2007; Kaplan et al, 2008a; 2008b; Roehrborn et al, 2009; Rovner et al, 2008a). This effect was obtained irrespective of prostate size and was not associated with increased incidence of acute urinary retention (Roehrborn et al, 2009). Darifenacin is a tertiary amine with moderate lipophilicity, well absorbed from the gastrointestinal tract after oral administration, and extensively metabolized in the liver by the cytochrome P450 isoforms CYP3A4 and CYP2D6, the latter saturating within the therapeutic range (Skerjanec 2006). UK-148,993, UK-73,689, and UK-88862 are the three main circulating darifenacin metabolites, of which only UK-148,993 is said to have significant antimuscarinic activity. However, available information suggests that various metabolites of darifenacin contribute little to its clinical effects (Michel and Hegde, 2006). The metabolism of darifenacin by CYP3A4 suggests that coadministration of a potent inhibitor of this enzyme (e.g., ketoconazole) may lead to an increase in the circulating concentration of darifenacin (Kerbusch et al, 2003). Darifenacin is a relatively selective muscarinic M3 receptor antagonist. In vitro, it is selective for human cloned muscarinic M3 receptors relative to M1, M2, M4, or M5 receptors. Theoretically, drugs with selectivity for the M3 receptor can be expected to have clinical efficacy in OAB/DO with reduction of the adverse events related to the blockade of other muscarinic receptor subtypes (Andersson, 2002b). However, the clinical efficacy and adverse effects of a drug are dependent not only on its profile of receptor affinity but also on its pharmacokinetics and on the importance of muscarinic receptors for a given organ function. Darifenacin has been developed as a controlled-release formulation, which allows once-daily dosing. Recommended dosages are 7.5 and 15 mg/day. The clinical effectiveness of the drug has been documented in several RCTs (Haab et al, 2004; Cardozo and Dixon 2005; Chapple et al, 2005, 2007a; Foote et al, 2005; Steers et al, 2005; Haab et al, 2006; Hill et al, 2006; Zinner et al, 2006; Abrams et al, 2008, Chancellor et al, 2008b; Dwyer et al, 2008; for reviews, see Guay, 2005; Zinner, 2007; Chapple et al, 2008; Novara et al, 2008). Haab and coworkers (2004) reported a multicenter, double-blind, placebo-controlled, parallel-group study that enrolled 561 patients (age 19 to 88; 85% female) with OAB symptoms for more than 6 months and included some patients with prior exposure to antimuscarinic agents. After washout and a 2-week placebo run-in, patients were randomized (1 : 4 : 2 : 3) to once-daily oral darifenacin controlled-release tablets: 3.75 mg (n = 53), 7.5 mg (n = 229) or 15 mg (n = 115) or matching placebo (n = 164) for 12 weeks. Patients recorded daily incontinence episodes, micturition frequency, bladder capacity (mean volume voided), frequency of urgency, severity of urgency, incontinence episodes resulting in change of clothing or pads, and nocturnal awakenings due to OAB using an electronic diary during weeks 2, 6, and 12 (directly preceding clinic visits). Tolerability data were evaluated from adverse event reports. Darifenacin 7.5 mg and 15 mg had a rapid onset of effect, with significant improvement compared with placebo being seen for most parameters at the first clinic visit (week 2). Darifenacin 7.5 mg and 15 mg were significantly superior to placebo for (median) improvements in micturition frequency (7.5 mg: −1.6; 15 mg: −1.7; placebo −0.8), frequency of urgency per day (−2.0; −2.0; −0.9), and number of incontinence episodes leading to a change in clothing or pads (−4.0; −4.7; −2.0). There was no significant reduction in nocturnal awakenings due to OAB. The most common adverse events were mild-to-moderate dry mouth and constipation with a CNS and cardiac safety profile comparable to that of placebo. No patients withdrew from the study as a result of dry mouth, and discontinuation related to constipation was rare (0.6% placebo vs. 0.9% darifenacin). In a dose titration study on 395 OAB patients, darifenacin, allowing individualized dosing (7.5 or 15 mg), was found to be effective and well tolerated (Steers et al, 2005). A 2-year open-label extension study of these investigations (i.e., Haab et al, 2004; Steers et al, 2005) confirmed a favorable efficacy tolerability and safety profile (Haab et al, 2006). A review of the pooled darifenacin data from the three phase 3, multicenter, double-blind clinical trials in patients with OAB was reported by Chapple and coworkers (2005). After a 4-week washout/run-in period, 1059 adults (85% female) with symptoms of OAB (urgency incontinence, urgency and frequency) for at least 6 months were randomized to once-daily oral treatment with darifenacin: 7.5 mg (n = 337) or 15 mg (n = 334) or matching placebo (n = 388) for 12 weeks. Efficacy was evaluated using electronic patient diaries that recorded incontinence episodes (including those resulting in a change of clothing or pads), frequency and severity of urgency, micturition frequency, and bladder capacity (volume voided). Safety was evaluated by analysis of treatment-related adverse events, withdrawal rates, and laboratory tests. Relative to baseline, 12 weeks of treatment with darifenacin resulted in a dose-related significant reduction in median number of incontinence episodes per week: 7.5 mg, −8.8 (−68.4%; placebo −54%, P < .004); 15 mg, −10.6 (−76.8%; placebo 58%, P < .001). Significant decreases in the frequency and severity of urgency, micturition frequency, and number of incontinence episodes resulting in a change of clothing or pads were also apparent, along with an increase in bladder capacity. Darifenacin was well tolerated. The most common treatment-related adverse events were dry mouth and constipation, although together these resulted in few discontinuations (7.5 mg, 0.6% of patients; 15 mg, 2.1%; placebo, 0.3%). The incidence of CNS and cardiovascular adverse events was comparable to that of placebo. The results were confirmed in other RCTs, including also a pooled analysis of three phase 3 studies in older patients (>65 years), showing that darifenacin (7.5 and 15 mg) had an excellent efficacy, tolerability, and safety profile (Foote et al, 2005; Zinner et al, 2005; Hill et al, 2006). One of the most noticeable clinical effects of antimuscarinic agents is their ability to reduce urgency and allow patients to postpone micturition. A study was conducted to assess the effect of darifenacin on the “warning time” associated with urinary urgency. This was a multicenter, randomized, double-blind, placebo-controlled study consisting of 2 weeks’ washout, 2 weeks’ medication-free run-in, and a 2-week treatment phase (Cardozo and Dixon, 2005). Warning time was defined as the time from the first sensation of urgency to voluntary micturition or incontinence and was recorded via an electronic event recorder at baseline (visit 3) and study end (visit 4) during a 6-hour clinic-based monitoring period, with the subject instructed to delay micturition for as long as possible. During each monitoring period, up to three urgency-void cycles were recorded. Of the 72 subjects who entered the study, 67 had warning time data recorded at both baseline and study end and were included in the primary efficacy analysis (32 on darifenacin, 35 on placebo). Darifenacin treatment resulted in a significant (P < .004) increase in mean warning time with a median increase of 4.3 minutes compared with placebo (darifenacin group, from 4.4 to 1.8 minutes; placebo, from 7.0 to −1.0 minute). Overall, 47% of darifenacin-treated subjects compared with 20% receiving placebo achieved a 30% or more increase in mean warning time. There were methodologic problems associated with this study; it utilized a dose of 30 mg (higher than the dose recommended for clinical use), the treatment period was short, it was conducted in a clinic-centered environment, the methodology carried with it a significant potential training effect, and the placebo group had higher baseline values than the treatment group. In another warning time study (Zinner et al, 2006) with 445 OAB patients, darifenacin treatment (15 mg) resulted in numerical increases in warning time; however, these were not significant compared with placebo. Further studies have demonstrated that darifenacin treatment is associated with clinically relevant improvements on health-related quality of life in patients with OAB (Abrams et al, 2008), and such improvements were sustained as shown in a 2-year extension study (Dwyer et al, 2008). It was shown that the positive effects neither on micturition variables nor on health-related quality of life produced by darifenacin (7.5 and 15 mg) were further enhanced by a behavioral modification program including timed voiding, dietary modifications, and Kegel exercises (Chancellor et al, 2008b). Several studies have been devoted to study possible effect on cognition by darifenacin. Neither in healthy volunteers (19 to 44 years) and healthy subjects (>60 years) nor in volunteers 65 years or older could any effect of darifenacin (3.75 to 15 mg/day) be demonstrated compared with placebo (Kay and Wesnes, 2005; Lipton et al, 2005; Kay et al, 2006; Kay and Ebinger, 2008). To study whether darifenacin had any effect on QT/QTc intervals, Serra and associates (2005) performed a 7-day, randomized, parallel-group study (n = 188) in healthy volunteers receiving once-daily darifenacin at steady-state therapeutic (15 mg) and supratherapeutic (75 mg) doses alongside control subjects receiving placebo or moxifloxacin (positive control, 400 mg) once daily. No significant increase in QTcF interval could be demonstrated compared with placebo. Mean changes from baseline at pharmacokinetic Tmax versus placebo were −0.4 and −2.2 msec in the darifenacin 15-mg and 75-mg groups, respectively, compared with +11.6 msec in the moxifloxacin group (P < .01). The conclusion was that darifenacin does not prolong the QT/QTc interval. Darifenacin 15 mg/day given to healthy volunteers did not change heart rate significantly compared with placebo (Olshansky et al, 2008). Darifenacin has a well-documented effect in OAB/DO, and the adverse event profile seems acceptable. Solifenacin is a tertiary amine and well absorbed from the gastrointestinal tract (absolute bioavailability 90%). The mean terminal half-life is 45 to 68 hours (Kuipers et al, 2004; Smulders et al, 2004). It undergoes hepatic metabolism involving the cytochrome P450 enzyme system (CYP3A4). In subjects who received a single oral dose of 10 mg solifenacin on day 7 of a 20-day regimen of ketoconazole administration (200 mg) Cmax and AUC0-inf were increased by only approximately 40% and 56%, respectively (Swart et al, 2006). Solifenacin has a modest selectivity for M3 over M2 receptors and a marginal selectivity over M1 receptors (Ikeda et al, 2002; Abrams and Andersson, 2007). Two large-scale phase 2 trials with parallel designs, comprising men and women, were performed (Smith et al, 2002; Chapple et al, 2004a). The first dose-ranging study evaluated solifenacin (2.5 mg, 5 mg, 10 mg, and 20 mg) and tolterodine (2 mg twice daily) in a multinational placebo-controlled study of 225 patients with urodynamically confirmed DO (Chapple et al, 2004a
Pharmacologic Therapy to Facilitate Bladder Filling and Urine Storage
Inhibiting Bladder Contractility, Decreasing Sensory Input, Increasing Bladder Capacity
Bladder Contraction and Muscarinic Receptors
Muscarinic Receptors
Drugs Used for Treatment of Detrusor Overactivity/Overactive Bladder Symptomatology
Levels of Evidence
Grades of Recommendation
ANTIMUSCARINIC DRUG
LEVEL OF EVIDENCE
GRADE OF RECOMMENDATION
Tolterodine
1
A
Trospium
1
A
Solifenacin
1
A
Darifenacin
1
A
Fesoterodine
1
A
Propantheline
2
B
Atropine, hyoscyamine
3
C
Drugs Acting on Membrane Channels
Calcium antagonists
2
D
Potassium channel openers
2
D
Drugs with Mixed Actions
Oxybutynin
1
A
Propiverine
1
A
Flavoxate
2
D
Antidepressants
Imipramine
3
C
Duloxetin
2
C
α-Adrenergic Receptor Antagonists
Alfuzosin
3
C
Doxazosin
3
C
Prazosin
3
C
Terazosin
3
C
Tamsulosin
3
C
β-Adrenergic Receptor Antagonists
Terbutaline (β2)
3
C
Salbutamol (β2)
3
C
YM178 (β3)
2
BPDE-
Phosphodiesterase-5 Inhibitors
Sildenafil, Tadalafil, Vardenafil
2
B
Cyclooxygenase Inhibitors
Indomethacin
C
Flurbiprofen
2
C
Toxins
Botulinum toxin (neurogenic)*
2
A
Botulinum toxin (idiopathic)*
3
B
Capsaicin†
2
C
Resiniferatoxin†
2
C
Other Drugs
Baclofen‡
3
C
Hormones
Estrogen
2
C
Desmopressin§
1
A
Antimuscarinic (Anticholinergic) Agents
Mechanism of Action
Pharmacologic Properties
REGION
SUBTYPE OF MUSCARINIC RECEPTOR
FUNCTION
Bladder
M2 > M3 (3 : 1)
M3: mediates human detrusor contraction
Salivary glands, parotid gland
M1, M3
M1: high viscosity lubrication
M3: salivation
Gastrointestinal tract
M2 > M3 (4 : 1)
M3: stimulation of gastrointestinal motility
Brain
M1 to M5 (M3 sparse)
Involved in higher cognitive processes such as learning and memory (mostly M1)
Eye
M1 to M5 (M3 predominates)
Controls iris sphincter contraction
Heart
M2
Modulates pacemaker activity, atrioventricular conduction, and the force of contraction
Clinical Efficacy
Tolerability and Safety
Atropine Sulfate
Propantheline Bromide
Trospium Chloride
Tolterodine Tartrate
Darifenacin Hydrobromide
Solifenacin Succinate
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree