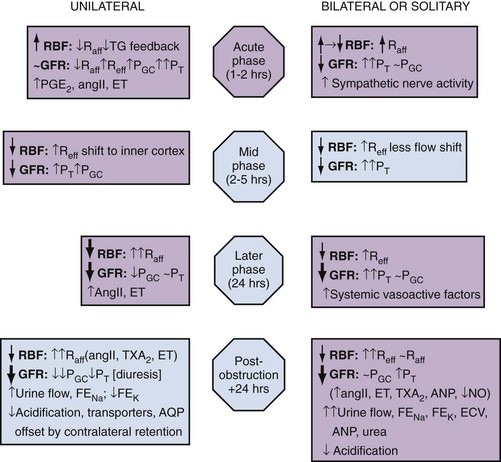
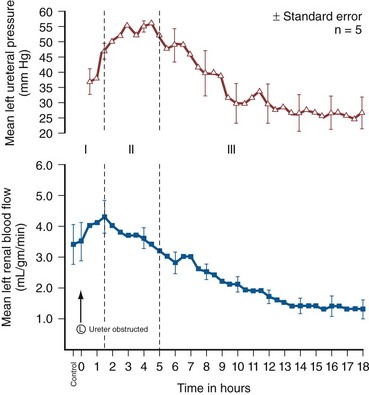
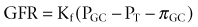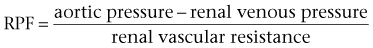Iqbal Singh, MCh (Urology), DNB (Genitourinary Surgery), MS, DNB, Jack W. Strandhoy, PhD, Dean G. Assimos, MD The prevalence of urinary tract obstruction is best estimated from autopsy series. In a series of 59,064 autopsies performed on individuals ranging from neonates to geriatric subjects, hydronephrosis was reported in 3.1% (Bell, 1950). There were no gender differences in this series until the age of 20 years. However, hydronephrosis was more prevalent in women in the 20- to 60-year interval. The latter was attributed to pregnancy and development of gynecologic malignancies. It was more prevalent in males after age 60 years, because of prostatic diseases. Hydronephrosis has been reported to be present in 2% to 2.5% of children subjected to autopsy (Campbell, 1970; Tan et al, 1994). It is somewhat more prevalent in boys, and the majority of cases were in subjects younger than 1 year. The aforementioned prevalences are most likely an underestimate of obstructive events, because temporary bouts of obstruction such as those induced by prior pregnancy or stone episodes were not captured. There are no new data regarding the prevalence and incidence of obstructive uropathy. However, there are new data on ureteropelvic junction obstruction and ureterocele that may permit certain inferences. Based on the United States published and coded data on ureteropelvic junction obstruction generated by the Healthcare Cost and Utilization Project (HCUP) and Kids’ Inpatient Database (KID), the number of inpatient hospitalizations for ureteropelvic junction have decreased from 1.1 per 100,000 in 1994 to 0.8 per 100,000 in 2000. Although this may be influenced by a shift to more outpatient procedures or observational management, it could also be due to a decreasing incidence of this problem (Schulam et al, 2007). However, this has not been demonstrated for those afflicted with ureterocele where the rates have been stable over this interval (Pohl et al, 2007). The pathophysiology of obstructive nephropathy is discussed in detail in the beginning of this chapter to provide a platform for understanding the clinical ramifications of this process, some of which are detailed in later sections that focus on unique causes of this problem. The mechanisms of obstructive nephropathy are unveiled at genetic, molecular, cellular, glomerular, renal tubular, whole kidney, and systemic levels. The impact of obstruction on the fetal and developing kidney is not emphasized because this is covered in other sections of this book. A list of possible causes of obstructive nephropathy is provided in Table 40–1. Table 40–1 Possible Causes of Obstructive Nephropathy Animal experiments have demonstrated a triphasic pattern of RBF and ureteral pressure changes in UUO that differs from BUO or unilateral obstruction of a solitary kidney (Fig. 40–1). With UUO, RBF increases during the first 1 to 2 hours and is accompanied by a high PT and collecting system pressure because of the obstruction. In a second phase lasting 3 to 4 hours, these pressure parameters remain elevated but RBF begins to decline. A third phase beginning about 5 hours after obstruction is characterized by a further decline in RBF, now paralleled by a decrease in PT and collecting system pressure. These changes are explained by physical alterations in flow dynamics within the kidney and are modified by changes in the biochemical and hormonal milieu regulating renal resistance. (From Moody TE, Vaughn ED Jr, Gillenwater JY. Relationship between renal blood flow and ureteral pressure during 18 hours of total unilateral ureteral occlusion. Implications for changing sites of increased renal resistance. Invest Urol 1975;13:246–51.) In the first phase of UUO, the increase in PT would logically be expected to reduce GFR greatly. However, this is counterbalanced by an increase in RBF related to afferent arteriolar vasodilation (Vaughan et al, 1970), which limits the fall in GFR because PGC rises. This hyperemic response has been attributed to stimulation of the tubuloglomerular feedback mechanism that relaxes the afferent arterioles as a consequence of decreased sodium delivery to the macula densa (Wright and Briggs, 1979), changes in interstitial pressure within the kidney (Vaughan et al, 1971; Francisco et al, 1980), or the release of vasodilators such as prostanoids like prostaglandin E2 (PGE2) (Allen et al, 1978). There are various lines of evidence supporting the role of prostaglandin involvement. Frøkiær and Sørensen (1995) demonstrated an increase in PGE2 excretion in the urine from the contralateral kidney after UUO. In addition, studies have shown that the increase in PGE2 and the vasodilation of the obstructed kidney could be blocked by indomethacin, a prostaglandin synthesis inhibitor (Allen et al, 1978; Blackshear and Wathen, 1978; Gaudio et al, 1980). Nitric oxide (NO) may also contribute to the early renal vasodilation in UUO. The kidney contains nitric oxide synthases (NOS) that are both constitutive (endothelial and neuronal isoforms) and inducible (iNOS). In obstructed kidneys from rabbits and rodents, iNOS increases (Salvemini et al, 1994; Miyajima et al, 2001). Furthermore, Lanzone and colleagues (1995) showed that administration of the NOS inhibitor NG-monomethyl-L-arginine (L-NMMA) before UUO attenuated the early rise in RBF, and that the renal vasodilation returned when L-NMMA was discontinued. Thus it is likely that both PGE2 and NO contribute to the net renal vasodilation that occurs early following UUO. After this initial phase of several hours, GFR and RBF progressively decline in UUO (Jaenike, 1972; Harris and Yarger, 1974; Dal Canton et al, 1980). In contrast to the early rise in PT in the initial phases of UUO, this parameter and RBF both decline 12 to 24 hours after obstruction. This is best explained by an increase in afferent arteriolar resistance (Raff) that occurs. At this time, there are also shifts in regional blood flow in the kidney, with large portions of the cortical vascular bed not perfused or underperfused (Harris and Yarger, 1974; Gaudio et al, 1980). A shift in RBF from the outer cortex to more juxtamedullary regions was reported by Yarger and Griffith (1974) in dogs with UUO. Thus reduced whole kidney GFR at this stage of obstruction is due not only to reduced perfusion of individual glomeruli, related to afferent vasoconstriction and reduced PGC, but also to global reduction in filtration related to no perfusion or underperfusion of many glomeruli (Arendshorst et al, 1974). Vasoconstrictors appear to play a role in the reduction in RBF after UUO. There is evidence that the renin-angiotensin system is activated during UUO, that is, during the first phase of UUO renal vein renin levels increase (Moody et al, 1975; Yarger et al, 1980) even though there is a net renal vasodilation at this time. Infusion of the angiotensin-converting enzyme (ACE) inhibitor captopril attenuates the declines in RBF and GFR in UUO, suggesting that angiotensin II is an important mediator of the preglomerular vasoconstriction occurring during the second and third phases of UUO (Ichikawa et al, 1985). Other vasoconstrictors also appear to be involved in the reduction of RBF with UUO. Thromboxane A2 (TXA2) is also thought to be an influential postobstructive vasoconstrictor that contributes to the continued reduction in GFR and RBF. Administration of TXA2 synthesis inhibitors to the obstructed kidney limits the reduction in RBF and GFR (Klotman et al, 1986; Loo et al, 1987; Purkerson and Klahr, 1989). TXA2 may be generated in the kidney itself, perhaps in glomeruli (Yanagisawa et al, 1990), but synthesis from macrophages migrating to the kidney during obstruction is another potential source of this vasoconstrictor (Schreiner et al, 1988; Harris et al, 1989). Endothelin is another endogenous vasoconstrictor thought to participate in these events, although perhaps later in the established phase of UUO and after release of the obstruction. Administration of endothelin antagonists limits the reduction of RBF and GFR in rats during and after release of UUO (Bhangdia et al, 1998; Syed et al, 1998; Colon et al, 2000). Furthermore, endothelin excretion is increased in the targeted kidney after release of UUO in swine but not in the contralateral renal unit (Kelleher et al, 1992). The kidney’s response to the release of UUO depends on the duration and extent of obstruction and is also species specific. Many models of UUO use complete obstruction of the ureter for 24 hours before release. After release of 24-hour UUO, the GFR is initially 50% of normal in dogs and less than 25% of normal in rats, accompanied by greatly reduced RBF. There are also regional differences within the kidney. Harris and Yarger (1974) showed a marked decrease in perfusion of the superficial cortex accompanied by an increase in juxtamedullary glomerular perfusion after release of 24 hours of UUO in rats. Tubuloglomerular feedback may play a role in these responses (Tanner, 1985). Some of the mediators of the hemodynamic changes in the kidney following release of the obstruction may be different from those involved in the earlier phases. The changes with BUO or obstruction of a solitary kidney are different. In contrast to the early robust renal vasodilation with UUO, there is a modest increase in RBF with BUO that lasts approximately 90 minutes, followed by a prolonged and profound decrease in RBF that is greater than found with UUO (Gulmi et al, 1995). Reyes and Klahr (1992) found that an NO synthesis antagonist caused a further decline in RBF and GFR compared with control values, suggesting that NO helps maintain renal hemodynamics in early BUO. Other potential vasodilators, such as platelet-activating factor (PAF), have been postulated to contribute to renal hemodynamic changes with BUO. Reyes and Klahr (1991) showed, in rats, that when vasoconstrictors such as TXA2 were blocked, endogenous or exogenous intrarenal PAF-mediated vasodilation contributed to the preservation of RBF and GFR. The earlier and more profound decrease in RBF with BUO may be contributed to by increased renal nerve stimulation that initiates vasoconstriction related to increased renorenal reflex activity (Francisco et al, 1980; Ma et al, 2002). Endothelin may also contribute to these responses in BUO. The administration of an endothelin antibody to rats with BUO was reported to attenuate the decreases in GFR and RPF (Reyes and Klahr, 1992). Angiotensin II and TXA2 are also probably involved in the changes occurring with BUO. The administration of inhibitors of either of these vasoconstrictors to rats, prior to BUO, resulted in improved postobstructive GFR and RPF compared with their administration at the time of release of obstruction (Purkerson and Klahr, 1989). The intrarenal distribution of blood flow is quite different with BUO than with models of UUO. Jaenike (1972) used microspheres to show that 55% of the RBF perfused the cortical nephrons, while the innermost zones received only 14% of the flow in rats after BUO. Similarly, Solez and associates (1976) showed a 92% decrease in inner medullary plasma flow with 18 hours of BUO in rats. Thus the shift seen with UUO of blood flow from outer to inner cortex is the opposite of that with BUO. Ureteral pressure is higher with BUO than with UUO. Although in both cases ureteral and tubular pressures are increased for the first 4 to 5 hours, the ureteral pressure remains elevated for at least 24 hours with BUO, whereas it begins to decline and approaches preocclusion pressures by 24 hours with UUO. The prolonged elevation in intratubular pressure contributes to the profound decrease in SNGFR and whole kidney GFR. Micropuncture studies (Yarger et al, 1972; Dal Canton et al, 1980) in which intratubular pressure is measured directly have demonstrated that it remains elevated in rats after 24 hours of BUO in comparison with pressure normalization in animals after 24 hours of UUO. Ureteral pressure remains high because BUO passes through a phase of preglomerular vasodilation and then a prolonged postglomerular vasoconstriction. This explains the persistent elevation in ureteral pressure in spite of a decrease in RBF and increase in renal resistance. In contrast, in UUO the initial preglomerular dilation and short-lived postglomerular vasoconstriction are followed by a more prolonged preglomerular vasoconstriction that tempers elevations in PGC and hence in PT. This difference between the two pathophysiologic conditions has been hypothesized to be due to an accumulation of vasoactive substances in BUO that could contribute to preglomerular vasodilation and postglomerular vasoconstriction. Such substances would not accumulate in UUO because they would be excreted by the contralateral kidney. Atrial natriuretic peptide (ANP) appears to be one of these substances (Purkerson et al, 1989). With excretory ability abrogated, BUO increases intravascular volume, as evidenced by an increase in pulmonary capillary wedge pressure and body weight, which serves as the stimulus for secretion of ANP. ANP increases afferent arteriolar dilation and efferent arteriolar vasoconstriction, thus increasing PGC. It also decreases the sensitivity of tubuloglomerular feedback, inhibits release of renin, and increases Kf (Fried et al, 1987; Brenner et al, 1990; Cogan, 1990). In addition, changes within the renal unit may contribute to elevated pelvic pressure. In both rats and humans, ureteral obstruction increases COX-2 in the proximal dilated ureter but not in the distal nondilated ureter. When the obstruction was released, the urinary concentration of PGE2 was elevated. Furthermore, a COX-2 selective inhibitor reduced pelvic pressure. Thus intrarenal PGE2 acting on ureteral smooth muscle may contribute directly to increased pelvic pressure after obstruction (Nørregaard et al, 2006). Glomerular filtration and RBF remain depressed after release of BUO. This is due to persistent vasoconstriction of the afferent arteriole (Jaenike, 1972; Moody et al, 1977). The decrease in RBF in the renal medulla remains more prominent than in the cortex (Solez et al, 1976). Tubuloglomerular feedback does not appear to contribute to this response as it does with UUO (Tanner, 1985). Urine flow and sodium excretion are increased after release of BUO. ANP appears to play a prominent role in this response based on its natriuretic properties and those previously reviewed. It may also have a protective effect. Ryndin and associates (2005) reported that the administration of ANP or phosphoramidon, an inhibitor of ANP degradation, to rats with BUO resulted in improved GFR and a more brisk diuresis and natriuresis after release of obstruction. In summary, both UUO and BUO involve increases in renal vascular resistances and increases in ureteral pressures. However, the timing and regulation of these changes differ (Fig. 40–2). With UUO, early renal vasodilation primarily mediated by prostaglandins and NO is followed by prolonged vasoconstriction and normalization of intratubular-ureteral pressure as the contralateral kidney contributes to fluid balance. With BUO, little early vasodilation is seen, and vasoconstriction is more profound. When the obstruction is released, the postobstructive diuresis is much greater with BUO because volume expansion, urea and other osmolytes, and secreted ANP contribute to a profound diuresis and natriuresis. Recovery of renal function in adult animals with partial obstruction has been studied as a potential guideline for clinical treatment. Leahy and coworkers (1989) studied dogs in which partial UUO with stents was studied for up to 60 days. Reversibility of renal function was estimated by creatinine clearance. Renal function became normal in dogs partially obstructed for 14 days. Animals partially obstructed for 28 days recovered 31% of function, and those partially obstructed for 60 days recovered only 8% of function. Casts of the microvasculature showed arteriolar constriction, supporting the concept that postobstructive renal dysfunction is influenced by vascular responses. Much less is known about the roles of vasoactive or inflammatory mediators in the partial ureteral obstruction model. As with acute occlusive models, administration of indomethacin or meclofenamate, both prostaglandin synthetase inhibitors, decreases GFR and increases arteriolar resistance (Ichikawa and Brenner, 1979) so that changes in eicosanoids are implicated in partial obstruction responses. Several investigators have shown that the renin-angiotensin-aldosterone axis is activated with partial obstruction, including studies in the fetus. Gobet and colleagues (1999) studied fetal sheep in utero and showed that partial obstruction of the bladder outlet increased renin messenger RNA (mRNA) after 2 weeks and increased the expression of renal AT2 receptors as well as the mRNA for transforming growth factor beta (TGF-β), a mediator of fibrosis. One could hypothesize that changes in angiotensin II may be responsible for much of the hemodynamic, fibrotic, and apoptotic changes in neonatal animals with partial obstruction, as has been shown in more acute models. Beharrie and coworkers (2004) examined weanling male rats that had been subjected to a partial UUO. The partial obstruction led to proteinuria, hyperuricemia, and increased solute excretion primarily from the unaffected contralateral kidney. A parallel group of rats treated with the ACE inhibitor enalapril was protected from these changes. This suggests that angiotensin is involved with functional tubular changes as well. Eskild-Jensen and colleagues (2002) examined partial UUO for up to 24 weeks in young, postnatal pigs. The number of glomeruli was 28% less in the obstructed kidney, yet function was just transiently reduced by the occlusion. Thus partial neonatal obstruction can impair nephrogenesis independently of renal functional decline, and these changes may depend upon species, stage of renal development, or degree of occlusion. More work is needed to define the variables. Various methods have been used to create partial obstruction. The models for preparing partial ureteral occlusions fall into three main categories. Intraluminal stents or catheters of varying sizes have been placed in the ureters of sheep (Abu-Zidan et al, 1999) and in dogs (Ryan and Fitzpatrick, 1987; Leahy et al, 1989) to restrict flow. In rats, or in growing animals, many investigators have used the technique of Ulm and Miller (1962), which involves splitting the psoas muscle longitudinally to form a groove into which the ureter is placed. This method has a potential advantage of increased constriction in proportion to the growth of the animal. Beharrie and colleagues (2004) used such a technique in young male weanling rats. A major problem with studies involving partial obstruction is the inability to determine reproducibly and accurately the degree of obstruction induced with these techniques. Thornhill and associates (2005) devised a method in neonatal rat pups of ligating a ureter along with a wire of a calibrated diameter that was then removed to make a partial and graded constriction of one ureter. When ureteral diameter was reduced by 70% to 75%, renal growth and number of glomeruli were reduced and fibrosis and pelvic dilation were proportionately increased. GFR was reduced by 80% after 28 days of partial UUO. Such models may offer new opportunities to model the clinical condition reproducibly. Although the normal flow of urine from the kidney through the urinary tract is compromised with obstruction, urine may still egress from the kidney. An example of this is extravasation at the calyceal fornix (pyelosinus) that occurs with acute obstruction, typically ureteral stones (Stenberg et al, 1988). Extravasation of urine into the venous (pyelovenous) and lymphatic system (pyelolymphatic) may also occur in this setting. In chronic obstruction, fluid is thought to exit into the renal venous system. Obstruction of one or both kidneys can have profound effects on sodium, potassium, and hydrogen excretion and mechanisms of urinary concentration and dilution. In the case of UUO, relatively normal function of the nonobstructed kidney partially offsets the reduced ability of the postobstructive kidney to reabsorb solutes and water. Postobstructive diuresis, something that is commonly encountered after reversal of BUO, occurs uncommonly after release of UUO, probably as a consequence of the contralateral renal unit’s functional capacities that are enhanced by an upregulation of ion transporters (Li et al, 2007). The eventual correction of abnormal renal excretory function depends upon the degree and duration of obstruction. Changes in tubule function after correction of UUO related to ureteropelvic junction or ureteral obstruction were characterized in 10 patients by Gillenwater and colleagues (1975). The mean period of obstruction in these cases was 12 months, and the range was from days to years. Functions of the normal and obstructed kidney were evaluated 1 week after relief of the obstruction. The GFR in the obstructed kidney was significantly less than that in the non-obstructed kidney (24 vs. 60 mL/min), and the urine osmolality and osmolar clearance were all significantly less in the postobstructed kidney. The similar osmolar clearances (volume of urine required to excrete urinary solute isosmotically) relative to GFR of the previously obstructed and unobstructed kidneys indicate that there is a true concentrating defect in the obstructed kidneys 1 week after relief of obstruction. In normal physiology, arginine vasopressin (AVP) is secreted into the bloodstream from the posterior pituitary gland in response to increased serum osmolality or a reduction in effective circulating volume. AVP binds to the V2 vasopressin receptor located on the basolateral surfaces of the collecting duct cells. This promotes G protein signaling, resulting in generation of cyclic adenosine monophosphate (cAMP). Generation of cAMP in turn activates protein kinase A, which stimulates the fusion of cytoplasmic vesicles containing aquaporin 2 (AQP2) with the apical membranes of the collecting duct cells. The fusion causes the normally watertight membrane to become water permeable. This promotes transcellular absorption of water through the AQP2 channels, which is transported through aquaporin 3 (AQP3) and aquaporin 4 (AQP4) channels located in the basolateral cell membrane into the interstitium. This sequence of events is driven by the osmotic gradient of sodium (Knoers, 2005). Another aquaporin, aquaporin 1 (AQP1), is abundant in renal proximal tubules, the thin descending limb of Henle, and the descending vasa recta in the kidney. It promotes urinary concentration through the countercurrent multiplier by facilitating water transport from the descending limb of Henle into the interstitium (King et al, 2001). The onset of concentration defects may develop soon after obstruction. Jaenike and Bray (1960) demonstrated a concentrating defect in the unilaterally obstructed kidney after only 6 minutes of ureteral obstruction. Development of vasopressin resistance has been hypothesized as a mechanism of this occurrence. However, various studies have demonstrated conflicting results concerning whether vasopressin resistance is present. Vascular changes may play a role. Even after only 18 hours of UUO, Solez and colleagues (1976) found a decrease in inner medullary plasma flow that increased when the occlusion was released. Necrosis of both the inner and outer medullae was present, indicating that ischemia may contribute to the development of a concentrating defect. Evidence from Li and coworkers (2001) demonstrated that the polyuria following the release of BUO correlates with a decreased expression of the aquaporin water channels AQP1, AQP2, and AQP3 in rats. Release of obstruction resulted in polyuria that gradually decreased over a 30-day period, even though urinary concentrating capacity remained significantly impaired. Expression of AQP2 and AQP3 became normal by 30 days after release, but the expression of AQP1 remained decreased. Jensen and coworkers (2006) examined changes in water channels after bilateral ureteral obstruction in rats. As expected, postobstructive polyuria with reduced urine osmolality was accompanied by decreased expressions of AQP1, AQP2, and AQP3 compared with control rats. The AT1 antagonist candesartan attenuated reductions in GFR, urine output, and AQP suppression, as well as COX-2 induction in the inner medulla. These findings suggest that angiotensin II influences, directly or indirectly, the postobstructive diuresis of BUO. A decrease in sodium transport in the nephron appears to play an additional prominent role in the decreased ability of the postobstructed kidney to concentrate urine. When UUO is released after an occlusion period of 24 hours, total urine excretion is normal to modestly increased despite increased fractional excretion of sodium (FENa) in the previously obstructed kidney. This is attributed to the contralateral kidney compensating for the sodium losses of its mate. However, with BUO, sodium and water excretions may be quite robust after release of obstruction. The FENa may be increased to as much as 20 times normal in this setting (Zeidel and Pirtskhalaishvili, 2004). Although ANP appears to play a role in sodium diuresis after release of BUO, it is unlikely to affect sodium transport defects associated with UUO. The latter is most likely due to selective cell membrane changes in the nephron that reduce the number and effectiveness of sodium transporters. Such changes may also occur with BUO. In spite of differential quantitative responses between UUO and BUO after release of the obstruction, the reabsorption defects in segmental nephron Na+ transport are similar. Micropuncture studies of kidneys from animals have been undertaken to assess these responses. These studies demonstrate normal to modestly enhanced isotonic volume flux (Jv) in superficial proximal convoluted tubules after release of UUO or BUO. On the other hand, sodium delivery to the loop of Henle in juxtamedullary nephrons, and to the first accessible portion of the inner medullary collecting duct, is substantially increased after release of both UUO and BUO, indicative of reduced sodium transport. Sonnenberg and Wilson (1976) even found evidence for net influx of sodium throughout the medullary collecting duct, which further contributed to an increased FENa. However, this was more prominent with BUO in their rat model. Studies in isolated perfused nephron segments have also provided knowledge about the impact of obstruction at the level of the nephron. They have shown normal isotonic reabsorption in the superficial proximal convoluted tubules from animals with either UUO or BUO, whereas Jv in proximal straight tubules from juxtamedullary nephrons was demonstrated to be impaired (Hanley and Davidson, 1982). Similarly, chloride reabsorption and transport-dependent oxygen consumption (QO2) from the medullary thick ascending limb of the Henle loop (MTAL) were severely impaired (Hanley and Davidson, 1982; Hwang et al, 1993b). Cell suspensions from nephron segments have been used to assess the effects of obstruction at a cellular level. Active transport of Na+ across cell membranes requires apical entry through selective Na+ transporters or channels and basolateral exit driven by sodium-potassium adenosine triphosphatase (Na+,K+-ATPase). Furthermore, adequate adenosine triphosphate (ATP) must be generated to drive these primary transport steps. Hwang and associates showed that the amount and activity of the apical Na+,K+,2Cl− cotransporter and bumetanide binding sites were reduced in cells isolated from the MTAL derived from obstructed rabbit kidneys. Basolateral transport is also affected in that ouabain-sensitive oxygen consumption, an index of Na+,K+-ATPase activity, has been shown to be reduced in cell suspensions from obstructed kidneys (Hwang et al, 1993a). A marked decrease in amiloride-sensitive oxygen consumption and Na+ entry in isolated cells from the inner medullary collecting ducts of obstructed rabbit kidneys reflects reduced activity of the apical Na channel (ENaC). In addition, ouabain-sensitive transport as measured by oxygen consumption and ATPase activity was shown to be reduced in cells from this portion of the nephron harvested from obstructed kidneys (Hwang et al, 1993a). Because cell suspension studies indicate that ATP generation is not the rate-limiting step underlying sodium transport dysfunction in this setting, the evidence points to downregulation of sodium transporters. This may be due to translational factors (reduction in mRNA for transporter synthesis) or post-translational processing of receptor proteins (Hwang et al, 1993a). The signals responsible for downregulation of transporter activity with obstruction have not been clearly defined, but a number have been hypothesized. Stasis of tubular fluid flow may be one of the signals. When urine flow is obstructed, upstream Na+ delivery to apical cell membranes slows so that the transmembrane gradient is reduced. This could then serve as the signal for the downregulation of transporter activity or expression resulting in reduced active Na+ transport across the basolateral cell membrane (Zeidel, 1993). Several studies indicate that this is a feasible mechanism. For example, ouabain-sensitive Na+,K+-ATPase activity is reduced in MTAL and collecting duct cells when mineralocorticoid activity is controlled and chronic furosemide or amiloride is given to reduce Na+ entry into tubule cells (Petty et al, 1981; Grossman and Hebert, 1988). Additional studies have explored this concept in established cell lines. When A6 cells, an established line of collecting duct cells, are grown on a permeable substrate, Na+ influx across the apical membrane through the ENaC Na+ channel occurs. When Na+ entry was blocked, this induced a decreased expression of β-subunit of the epithelial sodium channel in the apical membrane (Rokaw et al, 1996). Ischemia has also been proposed as a signal in this setting, where ischemia that accompanies the reduced perfusion of the kidney with obstruction can also be a mediator of reduced transporter expression. The reduction in major renal sodium transporters reported with ischemic models of renal failure supports this concept (Kwon et al, 2000). Other proposed downstream signals include changes in renal interstitial pressure and local generation of natriuretic substances. Thus substrate delivery may be a regulatory step in the expression of sodium and possibly other transporters. Intrarenal and extrarenal substances and hormones can also modulate sodium transport. The milieu of influential hormones may be substantially different between UUO and BUO. A number of investigators have shown that obstruction markedly increases the endogenous production of PGE2 in the renal medulla. Furthermore, supraphysiologic concentrations of PGE2 are well known to produce natriuresis (Strandhoy et al, 1974), and studies in isolated tubules and cell suspensions show that PGE2 inhibits Na reabsorption in the MTAL and throughout the collecting duct (CD). One mechanism of this natriuretic response may be that PGE2 reduces the amount of Na+,K+-ATPase at the basolateral membrane (Marver and Bernabe, 1992). PGE2 can also inhibit the tubular effects of vasopressin (Zook and Strandhoy, 1980), thereby contributing to the free water loss from the post-UUO kidney. The influence of other substances and hormones in the postobstructed UUO kidney is counterbalanced or minimized by the contralateral renal unit. Li and coworkers (2007) found that BUO caused a persistent decrease in the alpha and beta subunits of the epithelial Na channel (ENaC) three days after relief of obstruction. Similar changes in ENaC occurred following UUO but only in the obstructed kidney. The contralateral control kidney showed no changes in ENaC expression. Furthermore, Jensen and coworkers (2006) showed in rats that at 2 days following BUO there was reduced expressions of the Na phosphate cotransporter (NaPi-2), the loop Na+,K,2Cl− cotransporter (NKCC2) and the Na+/H+ exchanger (NHE3). These decreases in Na transporters were consistent with the postobstructive natriuresis. Candesartan partially reduced these changes and attenuated the natriuresis. Extracellular fluid volume may be greatly expanded with BUO. When the BUO is relieved, both intrarenal and extrarenal factors greatly enhance salt and water excretion so that a postobstructive diuresis is often seen. The normal physiologic consequences of extracellular fluid volume expansion ensue. These include a downregulation of sympathetic tone and the secretion of aldosterone and manifestation of the effects of ANP and probably other natriuretic factors. Levels of ANP are significantly elevated following BUO but not UUO (Purkerson and Klahr, 1989). The direct and indirect consequences of increased ANP levels include partial support of glomerular filtration, reduction of renin secretion and effects of angiotensin on transport, reduced aldosterone secretion, and direct inhibition of Na transport in the collecting duct (Brenner et al, 1990). Furthermore, BUO results in the accumulation of osmotic substances such as urea that can contribute to salt and water loss when the obstruction is relieved (Harris and Yarger, 1975, 1977). The FENa following relief of BUO is typically greater than that after UUO because BUO causes retention of Na, water, urea nitrogen, and other osmolar substances and increased production of ANP, all of which stimulate a profound natriuresis. Although Na transporters are similarly downregulated in the affected kidney in UUO, the contralateral kidney adequately compensates to maintain Na balance. Obstruction has a complex impact on renal potassium handling, depending upon the type of obstruction. Harris and Yarger (1975) reported that there is a decrease in K+ excretion, in proportion to the decrease in GFR, after release of a 24-hour period of UUO. This may be partially due to reduced delivery of Na+ to the distal nephron and a low volume flow rate that would minimize the transmembrane gradient for K+
Prevalence of the Problem
Renal
Congenital
Polycystic kidney
Renal cyst
Peripelvic cyst
Ureteropelvic junction obstruction
Neoplastic
Wilms tumor
Renal cell carcinoma
Transitional cell carcinoma of the collecting system
Multiple myeloma
Inflammatory
Tuberculosis
Echinococcus infection
Metabolic
Calculi
Miscellaneous
Sloughed papillae
Trauma
Renal artery aneurysm
Ureter
Congenital
Stricture
Ureterocele
Obstructing megaureter
Retrocaval ureter
Prune-belly syndrome
Neoplastic
Primary carcinoma of ureter
Metastatic carcinoma
Inflammatory
Tuberculosis
Amyloidosis
Schistosomiasis
Abscess
Ureteritis cystica
Endometriosis
Miscellaneous
Retroperitoneal fibrosis
Pelvic lipomatosis
Aortic aneurysm
Radiation therapy
Lymphocele
Trauma
Urinoma
Pregnancy
Radiofrequency ablation
Bladder and Urethra
Congenital
Posterior urethral valve
Phimosis
Hydrocolpos
Neoplastic
Bladder carcinoma
Prostate carcinoma
Carcinoma of urethra
Carcinoma of penis
Inflammatory
Prostatitis
Paraurethral abscess
Miscellaneous
Benign prostatic hypertrophy
Neurogenic bladder
Urethral stricture
Global Renal Functional Changes
Hemodynamic Changes with Unilateral Ureteral Occlusion
Hemodynamic Changes with Bilateral Ureteral Occlusion
Partial Ureteral Occlusion
Egress of Urine from the Kidney
Effects of Obstruction on Tubular Function
Urinary Concentrating Ability
Sodium Transport
Potassium Transport
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Pathophysiology of Urinary Tract Obstruction