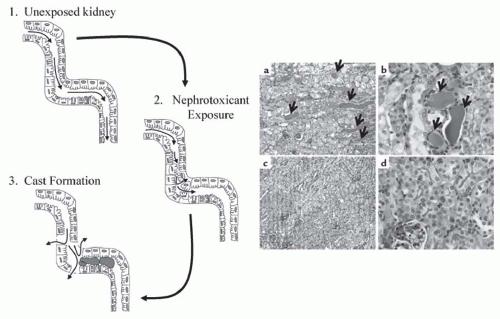
 FIGURE 30.1 Cast formation in the nephron. Left: 1.Filtrate flow (as represented by the small arrows) through the tubules is constant and unobstructed in unexposed kidneys. 2. Exposure of the kidney to nephrotoxicants results in cell injury and death, and can induce detachment of the cells from the basement membrane. 3.Detached cells can adhere to each other and form casts (pink), which obstruct filtrate flow and increase intraluminal pressure. This increases permeability in the basement membrane and back leak of filtrate into the interstitium. Right: Cisplatin-induced cast formation in wild-type mice (a and b) and mice mutant for tumor necrosis factor (TNF)-α (c and d) as determined by PAS staining. The magnification in panels a and c is ×100, and that in panels b and d is ×400. Cast formation is visible in panels a and b as indicated by the pink/purple aggregates between the tubules (arrows). In contrast, little cast formation can be seen in TNF-α knockout mice (panels c and d). (Adapted from Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest. 2002;110(6):835-842, with permission.) (See Color Plate.) |
the chemotherapeutics vincristine and doxorubicin. At least six different MRP genes have been identified (designated MRP1-6, and MDR1) and all are expressed in the kidney.49
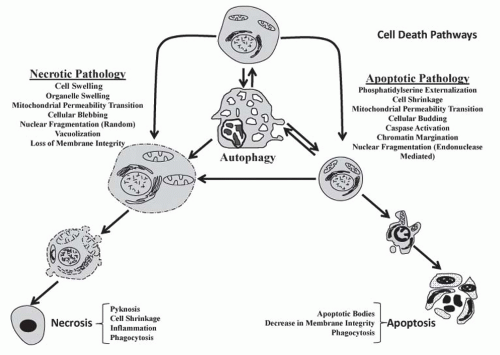 FIGURE 30.2 Schematic comparing the pathologic and morphologic features of necrosis, apoptosis, and autophagy. At the top middle, a normal cell is shown; below the normal cell is an autophagic cell demonstrating mass vacuolization; and autophagosomes (not shown). Left: Cell and organelle swelling, followed by vacuolization, blebbing, and increased membrane permeability (lysis) and finally necrotic changes (i.e., coagulation, shrinkage, and karyolysis). Right: Cell shrinkage followed by budding and karyorrhexis and finally necrotic changes (i.e., breakup into cluster of apoptotic bodies).The pathologies necrosis and apoptosis are listed. Double arrows represent the hypothesis that select pathways can switch. For example, autophagy can lead to cell survival and also progress to apoptosis. |
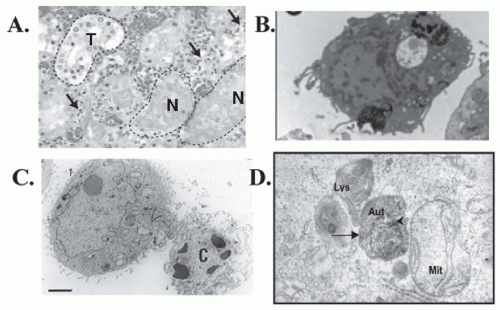 FIGURE 30.3 Comparison of the morphologic features of necrosis, apoptosis, and autophagy in tissues and cells. A: Hematoxylin and eosin staining of human kidney tissue after arterial embolization for treatment of renal cancer demonstrating intact tubules (T) and necrotic tubules (N). Arrows represent neutrophils and mononuclear inflammatory cells. (Modified from Hotchkiss RS, Strasser A, McDunn JE, et al. Cell death. N Engl J Med. 2009;361:1570, with permission.) B: Transmission electron microscopy (TEM) of necrotic human embryonic stem cells showing loss of membrane integrity without chromatin margination, cytosolic vacuolization, and spilling out of intracellular constituents. (Modified from Heng BC, Vinoth KJ, Lu K, et al. Prolonged exposure of human embryonic stem cells to heat shock induces necrotic cell death. Biocell. 2007;31(3):405, with permission.) C: TEM human HEK293 cells undergoing lysosomal mediated apoptosis. The cell on the right is relatively healthy whereas the cell on the left has shrunken and exhibits chromatin and nuclear condensation and is beginning to lose the membrane integrity (late apoptosis). (Modified from Heng BC, Vinoth KJ, Lu K, et al. Prolonged exposure of human embryonic stem cells to heat shock induces necrotic cell death. Biocell. 2007;31(3):405, with permission.) D: TEM of autophagic primary cultures of normal human renal cells exposed to cyclosporine demonstrating formation of the autophagosomes (Aut) with a double membrane (long arrow), next to a lysosomes (Lys) and a mitochondria (Mit).The arrowhead represents a cytoplasmic organelle. (Pallet N, Bouvier N, Legendre C, et al. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy. 2008;4(6):72,with permission.) |
(apoptotic body) or forms pseudopodia (i.e., buds) containing nuclear fragments and/or organelles that break off into small fragments (apoptotic bodies). In either case, adjacent cells or macrophages phagocytize the apoptotic bodies, and inflammation typically does not occur.
to apoptotic protease activating factor 1 (APAF-1), which promotes the binding and proteolytic cleavage of procaspase-9 to caspase-9 (the apoptosome),83 and then activated caspase-9 cleaves and activates executioner caspases (i.e., caspases-3, -6, and -7) (Fig. 30.4). Nephrotoxicants known to induce cytochrome c release in correlation with apoptosis include cisplatin and DCVC.71,84 Cytochrome c release from the mitochondria is associated with a decrease in the mitochondrial inner membrane potential and the accumulation of several pro-apoptotic proteins such as Bad, Bax, and Bax at the mitochondria (Fig. 30.4). Other proapoptotic proteins released from the mitochondria include apoptosis-inducing factor (AIF), Smac/Diablo, Omi, and Endo G (Fig. 30.4).70,71,85,86,87,88,89,90,91,92
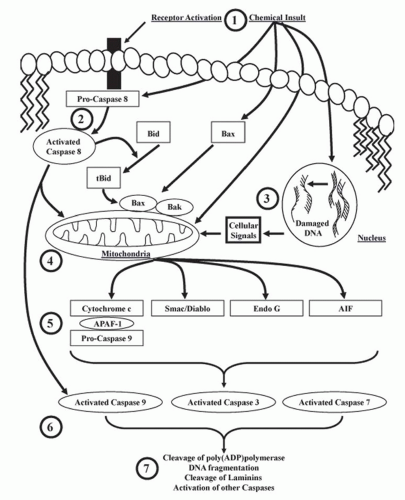 FIGURE 30.4 Cell signaling cascades involved in the activation of caspases and apoptosis. 1: Receptor-mediated death signals or chemicals can initiate apoptosis through multiple mechanisms. 2: Pro-caspase 8 is activated by receptor-mediated signals at the cellular membrane or directly by chemicals. Once activated, caspase-8 cleaves Bid to t-Bid, which interacts with Bax/Bak to induce mitochondrial-mediated apoptosis or directly activates caspase-9 and other caspases. 3: Some chemicals cause DNA damage that signals the release of pro-apoptotic proteins from the mitochondria. 4: Receptor-mediated signals, direct chemical injury, or signals resulting from DNA damage can all cause cytochrome c, Smac/Diablo, Endo G, and AIF release from the mitochondria. 5: Released cytochrome c forms a complex with APAF-1 and pro-caspase 9, resulting in caspase-9 activation. 6: Activated caspase-9 cleaves and activates procaspase-3 and -7, which can also be activated by caspase-8 independently of cytochrome c. 7: Activated caspases (e.g., 3 and 7), AIF, and Endo G cause the classical markers of apoptosis such as cleavage and activation of poly(ADP)polymerase, inactivation of inhibitors of DNases leading to DNA fragmentation, cleaved laminins, and the activation of other caspases. |
bioactivation, although renal cytochrome P-450 contributes to the nephrotoxicity of chloroform128,129 by metabolizing it to the unstable trichloroethanol, which releases HCl to form phosgene. Phosgene reacts with: (1) two molecules of glutathione to produce diglutathionyl dithiocarbonate, (2) water to produce two molecules of HCl and CO2, (3) cysteine to produce oxothizolidine-4-carboxylic acid, or (4) cellular macromolecules to initiate toxicity.128,130,131
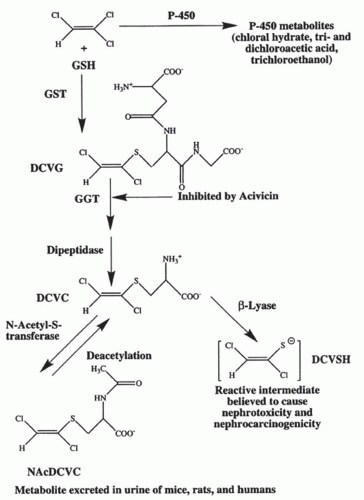 FIGURE 30.5 The bioactivation of trichloroethylene by the glutathione-(GSH-) conjugation pathway. Trichloroethylene (top left) can be metabolized by either cytochrome P-450 to the compound listed (top right) or be conjugated to GSH by the glutathione S-transferase (GST) to form S-(1,2)-dichlorovinyl-glutathione (DCVG).These reactions can occur either in the liver or in the kidney. DCVG formed in the liver is delivered to the kidney via the bile or the blood where the high concentrations of γ-glutamyltransferase (GGT) and dipeptidase in the kidney results in the cleavage of the GSH moiety and the formation of S-(1,2)-dichlorovinyl-L cysteine (DCVC). Metabolism of DCVC by N-acetyl-s-transferase produces N-acetyl-s-(1,2)-dichlorovinyl-L-cysteine (NAcDCVC), which is excreted in the urine of mice, rats, and humans exposed to trichloroethylene. NAcDCVC also can be deacetylated back to DCVC. Metabolism of DCVC by cysteine-conjugate β-lyase results in the formation of a reactive thiol that can rearrange to form a protein acylating species. (From Cummings BS, Parker JC, Lash LH. Role of cytochrome P450 and glutathione S-transferase alpha in the metabolism and cytotoxicity of trichloroethylene in rat kidney. Biochem Pharmacol. 2000;59:531, with permission.) |
or the release of the mitochondrial pro-apoptotic protein Smac/Diablo.
TABLE 30.1 Expression of Selected Xenobiotic Biotransformation Enzymes in the Kidney | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
suggest that sex- and age-dependent differences exist for the expression of FMO mRNA in the kidney142; however, no differences in the expression of FMO1, FMO3, or FMO5, and overall FMO activity were detected between human male and female kidney microsomes.141 Thus, more work is needed to determine if FMO expression is sex-dependent in human kidneys.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


