The earliest signs of clinical AKI are urinary muddy brown casts and an increased fractional excretion of sodium (FE
Na).
82,
83 The proximal tubule is the most frequent morphologic site of injury in ischemic AKI in both humans and animals.
84 The S
3 segment of the proximal tubule is particularly prone to ischemic injury, perhaps because of its location in the outer medulla, which is relatively hypoxic compared to the renal cortex.
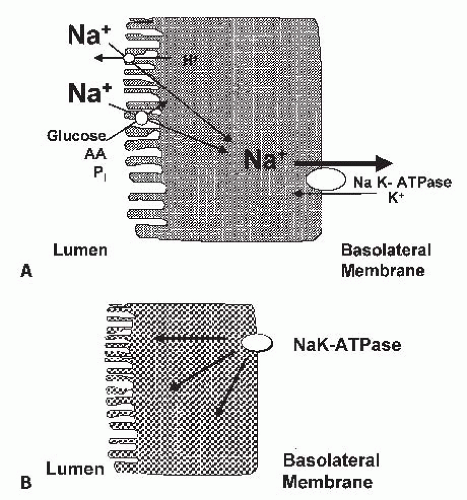
84 The proximal tubule nephron site is also associated with impaired vectorial sodium transport. The earliest morphologic changes with ischemic injury include invagination and sloughing of the brush border membrane into the lumen, an abnormality that is compatible with impairment and a loss of apical sodium antiporters and cotransporters responsible for sodium entry into the proximal tubular epithelium.
85,
86,
87 The tubules lose their polarity.
83 In vitro studies have shown that ATP depletion leads to dephosphorylation and inactivation of the actin binding protein, ezrin, and activation of the actin depolarizing protein in the proximal tubule membrane.
88,
89 This leads to a disruption of the microvillar actin and a loss of the brush border membrane.
90 A loss of polarity of the proximal tubule cells during chemical anoxia and ischemia has also been shown with the translocation of the Na-K-ATPase to the apical membrane (
Fig. 29.2B).
91,
92,
93 The translocated Na-K-ATPase remains functional.
94 In MDCK cells exposed to ATP depletion, there is a loss of polarity of Na-K-ATPase and a dissociation of the membrane-cytoskeleton complex at the spectrin-ankyrin interface.
95 Thus, sodium transport back
across the apical membrane and into the proximal lumen, as well as decreased proximal tubular sodium reabsorption, has been proposed in response to hypoxia and ischemia.
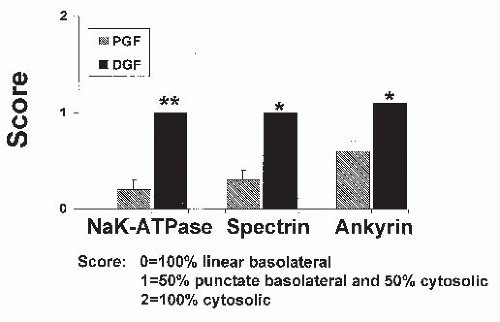
Studies in cadaveric transplanted kidneys with both prompt and delayed graft functions have been compared relative to the cellular location of the actin binding proteins, ankyrin and spectrin, and Na-K-ATPase using selective antibodies. In kidneys with delayed graft function, approximately 50% of the ankyrin, spectrin, and Na-K-ATPase were translocated from the basolateral membrane to the cytoplasm (
Fig. 29.3). Those kidneys with prompt graft function had only minimal translocation of these proteins from the basolateral membrane (
Fig. 29.3).
55 These observations therefore have contributed to our understanding of reversible and sublethal tubular dysfunction in ischemic kidneys in vivo (
Fig. 29.3).
Because proximal tubular cells in a culture convert from primarily oxidative to glycolytic metabolism and alter their phenotype,
34,
35 confirmation of experimental results in other systems is advisable. The use of freshly isolated proximal tubules to study the response to hypoxia has also been enlightening. These tubules, in general, maintain their phenotype and oxidative metabolism but are more sensitive to hypoxia, as assessed by LDH release, than in vivo tubules.
96,
97,
98,
99 Hypoxia for 15 to 30 minutes causes a reproducible release of LDH, and the cells die by necrosis.
48,
49 The central role of intracellular calcium and various protective maneuvers against hypoxic injury have been demonstrated in these isolated proximal tubules. However, before discussing the role of intracellular calcium in proximal tubular injury, we shall briefly consider adenine nucleotides. It is unquestioned that the first effect of ischemia, hypoxia, or mitochondrial inhibition in most in vitro and in vivo models is to compromise adenine nucleotide metabolism. A decreased production of ATP precedes the increase in intracellular calcium.
Adenine Nucleotides
The removal of oxygen from renal cells or whole kidneys results in prompt decreases in the cellular ATP pool. Initially, adenosine diphosphate (ADP) and adenosine monophosphate (AMP) concentrations increase,
52 and further catabolism of AMP to adenosine and then to hypoxanthine and, in some species to xanthine, occurs as the ischemic period is prolonged.
100,
101 The provision of exogenous ATP-MgCl
2 to ischemic rat kidneys protects against ischemic injury.
102 Mechanisms whereby a loss of ATP results in cellular injury include the loss of purine nucleosides themselves (in some species, the generation of oxygen free radicals during reperfusion), and the loss of many metabolic functions (e.g., phosphorylation of important enzymes, ion channels, and the functions of ion transporters that are dependent on adequate ATP levels).
Ischemic preconditioning protects the heart, and in some studies the kidneys, from subsequent ischemia-reperfusion injury. Ischemic preconditioning appears to be mediated via the activation of adenosine receptors, specifically the A
1 adenosine receptors. In support of this are studies that the exogenous administration of adenosine or A
1 adenosine agonists mimic ischemic preconditioning in cardiac muscle.
103 It was recently demonstrated that rat kidneys can be preconditioned to attenuate ischemic-reperfusion injury. In this study, adenosine infusion before the ischemic insult protects renal function via A
1 adenosine receptor activation, and adenosine A
1 antagonism blocks adenosine-induced protection. In a more recent study, acute and delayed protection against renal ischemia was seen with an A
1 adenosine receptor agonist.
104 In addition, adenosine A
3 receptor activation before the ischemia worsens the renal ischemia-reperfusion injury and A
3 receptor antagonism protects renal function.
103 A
2A adenosine receptors mediate the inhibition of ischemic AKI in rats due to an inhibitory effect on neutrophil adhesion.
105,
106 A combined infusion of an A
2A adenosine receptor agonist and a type IV phosphodiesterase (PDE 4) inhibitor leads to enhanced protection against ischemia-reperfusion injury in mice.
107 Protection against renal ischemia-reperfusion injury by A
2A receptor agonists or endogenous adenosine requires the activation of receptors expressed on bone marrowderived cells.
108 The A
2A adenosine receptor may be a novel therapeutic target in renal ischemia-reperfusion injury.
109,
110
Intracellular Calcium
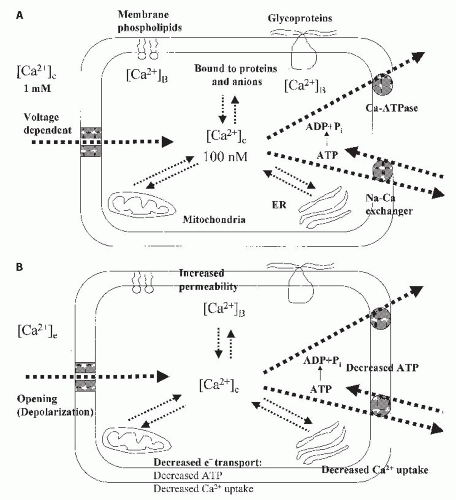
The normal regulation of epithelial cell calcium is demonstrated in
Figure 29.4A. Calcium exists in the cell as cytosolic free calcium, which is the smallest pool, but the most critical for regulation of intracellular events. Calcium is also bound to proteins and anions in the cytosol and to membrane
phospholipids and glycoproteins. The largest pools of intracellular calcium are in mitochondria and the endoplasmic reticulum.
111,
112 The concentration of calcium in the cytosol is about 100 nM, which is 1/10,000 of extracellular calcium concentrations.
111 The large electrochemical gradient between intracellular and extracellular calcium is maintained by the binding of calcium to intracellular components and by apical and basolateral transport systems. Transport systems may be voltage dependent or ATP dependent. Calcium efflux is mediated in basolateral membranes by both calcium ATPase, which is ATP dependent, and by a Na
+/Ca
2+ exchanger on the basolateral membrane, which is ATP independent.
113 Normally, the cell membrane is impermeable to calcium and maintains the steep calcium gradient between cytosolic free calcium and the extracellular space.
112 However, when cytosolic calcium increases in response to either increased cellular membrane permeability or decreased calcium efflux, or both, the mitochondria and the endoplasmic reticulum (ER)
actively increase their calcium uptake. Mitochondrial uptake and retention of calcium becomes substantial only when cytosolic levels exceed 400 to 500 nM, as occurs with cell injury.
111 Mitochondrial uptake is regulated by a calcium uniporter in the mitochondrial inner membrane. Thus, during cell injury, active mitochondrial sequestration appears to be quantitatively the most important process for buffering elevations in cytosolic calcium.
During epithelial cell injury, several factors favor increases in cytosolic free calcium (
Fig. 29.4B). This includes (1) decreased mitochondrial electron transport leading to decreased ATP levels, (2) increased membrane permeability, and (3) depolarization or opening of voltage-dependent channels. The decreased ATP leads to a decreased calcium uptake by mitochondria and ER and a decreased ability to pump calcium out of the cell.
With this background on the normal regulation of cell calcium, we shall now consider the role of intracellular calcium in tubular injury. In 1981, it was proposed that calcium ions were important participants in the functional, biochemical, and morphologic disturbances that characterize AKI.
114,
115 Numerous studies over the past 15 years in different injury models and cell types have demonstrated an increase in cytosolic calcium in renal epithelial cell injury. These studies are summarized in
Table 29.3.
The crucial questions to implicate calcium as a primary factor in cell injury are (1) whether the increase in cytosolic calcium precedes the injury and (2) whether preventing the rise in cytosolic calcium attenuates the injury.
116,
117 To investigate whether hypoxia is associated with an increase in free cytosolic calcium in proximal tubular cells, which precedes any evidence of membrane damage, a video imaging technique was developed in which free intracellular calcium could be measured simultaneously with staining of nuclei with the membrane impermeable indicator, propidium iodide, as an index of hypoxia-induced membrane damage.
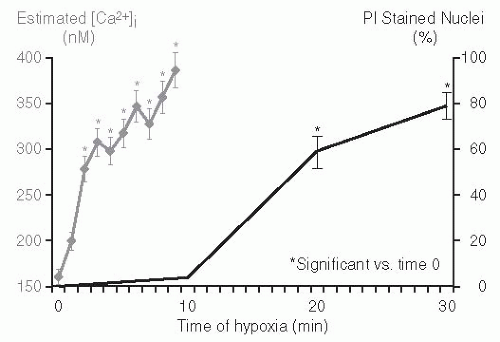
118 Propidium iodide enters the cell through the damaged plasma membrane and stains the cell nucleus. The percent of nuclei that stain with propidium iodide is quantitated and is an index of plasma membrane damage. Hypoxia in rat proximal tubules is associated with a significant rise in cytosolic calcium, which antecedes evidence of membrane damage as assessed by propidium iodide staining.
99 Cytosolic calcium increased from 170 to 390 nM during 5 minutes of hypoxia. The increase in cytosolic calcium preceded propidium iodide-detectable cell injury (
Fig. 29.5). The increase in cytosolic calcium that preceded the hypoxic membrane damage was promptly reversible with reoxygenation after 8 minutes of hypoxia. This is important because if cytosolic calcium is increased only after lethal cell membrane damage, reoxygenation should not have
normalized cytosolic calcium. The 10 minutes of cytosolic calcium rise correlated significantly with subsequent cell damage observed at the 20 minute mark. The pivotal role of the rise in cytosolic calcium during hypoxia was further demonstrated by using the intracellular Ca
2+ chelator BAPTA to prevent the rise in cytosolic calcium; this approach resulted in marked cytoprotection against hypoxic tubular injury.
What are the mechanisms whereby increases in cytosolic free calcium could lead to cell membrane injury? Potential calcium-dependent mechanisms include changes in the actin cytoskeleton of proximal tubule microvilli,
119 activation of calcium-dependent PLA
2,
40 and activation of the calcium-dependent cysteine protease, calpain.
116,
117,
120,
121
Calcium-Dependent Changes in the Actin Cytoskeleton
The role of calcium in pathophysiologic alterations of the proximal tubule microvillus actin cytoskeleton was studied in freshly isolated tubules.
119 Precisely defined medium calcium levels were defined using a combination of the metabolic inhibitor, antimycin, and the ionophore, ionomycin, in the presence of glycine to prevent lethal membrane damage. Increases of intracellular calcium to 10 µM were sufficient to initiate concurrent actin depolymerization, fragmentation of F-actin into forms requiring high-speed centrifugation for recovery, redistribution of villin to sedimentable fractions, and structural microvillar damage consisting of severe swelling and fragmentation of actin cores. However, during ATP depletion induced by antimycin alone or hypoxia alone, initial microvillar damage was calcium independent. This study suggests that both ATP depletion-dependent but Ca
2+-independent, as well as Ca
2+-mediated processes, can disrupt the actin cytoskeleton during acute proximal tubule cell injury; that both types of change occur, despite protection afforded by glycine and reduced pH against lethal membrane damage; and that Ca
2+-independent processes primarily account for prelethal actin cytoskeletal alterations during simple ATP depletion of proximal tubule cells.
In normal proximal tubule cells, actin is concentrated in apical brush border microvilli, along with the actin-binding protein, villin. Villin plays an important role in actin bundling and in microvillar assembly but can also act as an actin-fragmenting protein at higher calcium concentrations. The effects of ischemic injury and reperfusion on the distribution of villin and actin in the proximal tubule cells of rat kidneys were examined.
122 This study demonstrated that villin may be involved in the initial disruption of the actin cytoskeleton during reperfusion injury and that its migration back to the apical domain of these cells accompanies the reestablishment of a normal actin distribution in the brush border.
ATP depletion results in the conversion of monomeric G-actin to polymeric F-actin during tissue ischemia.
123 This conversion results from altering the ratio of ATP-G actin and ADP-G actin, causing a net decrease in the concentration of thymosin actin complexes as a cosequence of the differential affinity of thymosin beta 4 for ATP and ADP-G actin.
124 Recent studies suggest that the actin-binding protein tropomyosin binds to and stabilizes the apical actin microvilli under physiologic conditions in proximal tubules.
125
Activation of Phospholipase A2
Phospholipase A
2 (PLA
2) enzymes are important regulators of prostaglandin and leukotriene synthesis and can directly modify the composition of cellular membranes.
126 PLA
2 enzymes are also potent regulators of inflammation. The cytosolic form, cPLA
2, preferentially releases arachidonic acid from phospholipids and is regulated by changes in intracellular calcium concentration.
127PLA
2 enzymatic activity was measured in cell-free extracts prepared from rat renal proximal tubules.
40 Both soluble and membrane-associated PLA
2 activity were detected. All PLA
2 activity detected during normoxia was calcium dependent. The fractionation of cytosolic extracts by gel filtration revealed three peaks of PLA
2 activity. Exposure of tubules to hypoxia resulted in stable activation of soluble PLA
2 activity, which correlated with the disappearance of the highest molecular mass form (>100 kDa) and the appearance of a low-molecular-mass form (approximately 15 kDa) of PLA
2. Hypoxia also resulted in the release of a low-molecular-mass form of PLA
2 into the extracellular medium. Pretreatment of tubules with glycine before hypoxia blocked this release of PLA
2 but not the activation of soluble PLA
2 activity. This study provides direct evidence for calcium-dependent PLA
2 activation during hypoxia. However, calcium-independent forms of PLA
2 have also been found to play a role in hypoxic proximal tubular injury.
128The mechanism of PLA
2-induced cell membrane damage is interesting. Membrane phospholipid breakdown has been observed to occur in a number of tissues during ischemia.
129 In proximal tubules, hypoxia has been shown to
cause an increase in free fatty acids, which was initially believed to contribute to cell injury.
130 However, a study from our laboratory has shown that unsaturated free fatty acids protect against hypoxic injury in proximal tubules and that this protection may be mediated by a negative feedback inhibition of PLA
2 activity.
41 This protective effect of unsaturated free fatty acids has been confirmed by Zager et al.
131 The injurious effect of PLA
2 could be related to a direct disruption of cell membrane integrity by attacking the phospholipid component of cell membranes or through the accumulation of lysophospholipids, which have been shown to disrupt cell membranes and cause cytotoxicity.
132
Activation of Calpain
The cysteine proteases are a group of intracellular proteases that have a cysteine residue at their active site. The cysteine proteases consist of three major groups: cathepsins, calpains, and the newly discovered caspases. The major groups of cysteine proteases are shown in
Table 29.4. The cathepsins are non-calcium-dependent lysosomal proteases that do not appear to play a role in lethal cell injury.
133,
134,
135 Calpain is a calcium-activated neutral protease (CANP).
136 It has absolute dependence on calcium. There are two major ubiquitous or conventional isoforms of calpain, the low calcium sensitive µ-calpain and the high calcium sensitive m-calpain.
137,
138 The isoenzymes have the same substrate specificity but differ in affinity for Ca
2+. µ-Calpain is activated by micromolar concentrations of Ca
2+, and m-calpain is activated by millimolar concentrations of Ca
2+. The millimolar concentrations of intracellular calcium needed for the activation of m-calpain are not seen in normal cells, and phosphatidylinositol is thought to lower the calcium concentration required for half the maximal autolysis of m-calpain.
139 Procalpain exists in the cytoplasm as an inactive proenzyme and becomes active proteolytically only after it has become autolysed at the cell membrane. Activity of the autolysed calpain is subject to a final regulation by calpastatin.
140,
141 Calpastatin is a specific endogenous inhibitor of calpain. It is as widely distributed in nature as the enzyme itself. Calcium is required for calpastatin to bind to calpain and thus for the inhibitory effect of calpastatin on calpain.
Postulated functions of calpain include platelet activation and aggregation, cytoskeleton and cell-membrane organization,
142 and the regulation of cell growth.
143,
144,
145,
146The calcium-dependent calpains have been shown to be mediators of hypoxic/ischemic injury to the brain, liver, and the heart.
147,
148,
149,
150 The role of the calcium-dependent cytosolic protease, calpain, in hypoxia-induced renal proximal tubular injury has also been demonstrated.
43 Tubular calpain activity increased significantly by 7.5 minutes of hypoxia, before there was significant LDH release, and further increased during 20 minutes of hypoxia. Chemically dissimilar cysteine protease inhibitors markedly decreased LDH release after 20 minutes of hypoxia and completely prevented the rise in calpain activity during hypoxia. This role of calpain in proximal tubule injury has subsequently been confirmed by other groups.
151,
152 This increased calpain activity has subsequently been shown to be associated with a breakdown of the cytoskeletal protein, spectrin, both in vitro
45 and in vivo,
153 as well as increasing Na-K-ATPase into the cytoplasmic fraction of the cell.
Recent studies have demonstrated that calpain mediates progressive plasma membrane permeability and the proteolysis of cytoskeleton-associated paxillin, talin, and vinculin during antimycin A or hypoxia-induced proximal tubular cell death.
154 Novel nonpeptide calpain inhibitors are protective against antimycin A-induced calcium influx and hypoxia/reoxygenation-induced proximal tubular cell death.
155 In novel in vivo studies, calpastatin transgenic mice that had a decreased activation of calpain in the kidney
were generated.
156 In an anti-glomerular basement membrane (GBM) model, calpastatin-transgenic mice had less severe glomerular injury and a reduction in nuclear factor kappa-B (NF-κB) activation, suggesting a role for calpain in inflammation.
Caspases
Caspases are another group of intracellular cysteine proteases. Caspases participate in two distinct signaling pathways: (1) the activation of proinflammatory cytokines and (2) the promotion of apoptotic cell death.
9,
157,
158,
159,
160,
161,
162 Caspases 3 and 7 are the major mediators of apoptosis. The term “caspase” embodies two properties of these cysteine proteases in which “c” refers to “cysteine” and “aspase” refers to their specific ability to cleave substrates after an aspartate residue. The members of the caspase family are divided into subfamilies based on substrate specificity and function.
163 Caspase-1 (previously known as IL-1—converting enzyme [ICE]) plays a major role in the activation of proinflammatory cytokines. For many years it was not known how caspase-1 was activated. It has recently been discovered that procaspase-1 is activated in a complex called the inflammasome.
164,
165 The inflammasome is a protein scaffold that contains NALP (NACHT, LRR, and pyrin domain—containing) proteins, an adaptor protein called ASC (apoptosis-associated specklike protein containing a caspase-recruiting domain [CARD]), procaspase-1, and caspase-5. The interaction of the CARD of procaspase-1 is mediated by the CARD of ASC and the CARD present in the C-terminus of NALP-1. Active caspase-1 in the inflammasome is a regulator of the unconventional protein secretion of leaderless proteins like IL-1α and fibroblast growth factor (FGF)-2.
166 In a recent study, the inflammasome components NLRP3 and ASC were highly expressed in the renal tubular epithelium of humans and mice.
167 The absence of Nlrp3, but not ASC, protected against ischemic AKI.
167 Activation of caspases-1, 8, 9, and 3 have been described in hypoxic renal epithelial cells
168,
169,
170 and cerebral ischemia.
171 Although cells contain many caspases, the targeted disruption of specific caspase genes in mice has provided much insight into the functions of individual caspases during cell death.
172Although caspases play a crucial and extensively studied role in apoptosis, there is now considerable evidence that the caspase pathway may also be involved in necrotic cell death.
173 The inhibition of caspases protects against necrotic cell death induced by the mitochondrial inhibitor, antimycin A, in PC12 cells, Hep G2 cells, and renal tubules in a culture.
174,
175 Caspases are also involved in hypoxic and reperfusion injury in cultured endothelial cells.
176 Rat kidneys subjected to ischemia demonstrate an increase in both caspase-1 and caspase-3 mRNA and protein expression.
177 Caspases play a role in hypoxia-induced necrotic injury of isolated rat renal proximal tubules.
48 In this study, caspase activity was increased in association with cell membrane damage as assessed by LDH release. A specific caspase inhibitor attenuated the increase in caspase activity and markedly protected against cell membrane damage. To specifically identify the caspase involved in proximal tubular injury, proximal tubules were isolated from capsase-1 knockout mice and exposed to hypoxia.
178 Proximal tubules from caspase-1 knockout mice were protected against hypoxic injury, demonstrating the role of caspase-1 in directly causing cell membrane damage in proximal tubules.
178A study
179 investigated the role of caspase inhibition and apoptosis in ischemic AKI in mice in vivo. A relationship between apoptosis and subsequent inflammation was found. At the time of reperfusion, administration of the antiapoptotic agents insulin-like growth factor 1 (IGF-1) and ZVAD-fmk (a caspase inactivator) prevented the early onset of not only renal apoptosis, but also inflammation and tissue injury. Conversely, when the antiapoptotic agents were administered after the onset of apoptosis, these protective effects were completely abrogated.
There appears to be an interaction between caspases and calpain during hypoxia-induced injury in the proximal tubule, because caspase inhibition was shown to decrease calpain activity during hypoxia.
48,
121 Recent in vivo studies
153 suggest that caspase-mediated degradation of the endogenous inhibitor of calpain, calpastatin, is a mechanism whereby the calcium-mediated activity of calpain is increased.
Caspases in Cold Ischemia
Preservation injury, also known as cold ischemia, is an important clinical problem in kidney transplantation. Significant damage to the kidney may occur during harvest, cold storage, and transport. An ongoing area of interest is identifying methods to reduce organ injury during this process. The primary consequence of cold ischemic injury is delayed graft function (DGF) in kidney transplants.
180,
181,
182 These consequences have both short-term and long-term effects. In a kidney transplant, for example, DGF increases patient morbidity in the short term because the hospital stay is longer and dialysis may be required. In the long term, DGF independently predicts reduced 1- and 5-year graft survival.
183Both human and animal studies suggest that the adverse impact of cold ischemia may be associated with apoptosis. In human kidney transplant biopsies performed after 1 hour of reperfusion, apoptosis of tubular cells correlated significantly with cold ischemic time.
184 Biopsies of human donor kidneys, which subsequently developed postoperative AKI, demonstrated increased renal tubular epithelial cell apoptosis.
185 Prolonged cold ischemia has also been shown to increase apoptotic cell death in rat kidney allografts at 24 weeks posttransplant.
186 Mitochondria undergo significant changes during ischemia and may contribute to preservation injury.
187Caspases have been studied in cold ischemic kidneys.
188 Kidneys were stored for 48 hours at 4°C to produce cold ischemia. Caspase-3 activity was massively increased (100-fold) in cold ischemic kidneys compared to controls. On immunoblot analysis, the processed form of caspase-3
was increased in cold ischemic kidneys compared to controls. The increase in caspase-3 was associated with significantly more renal tubular apoptosis and brush border injury. The pan-caspase inhibitor prevented the formation of the processed form of caspase-3 and the increase in caspase activity, and reduced apoptosis and brush border injury. The results of this study suggest that caspase inhibition may prove useful in kidney preservation.
In a pig model of cold ischemia in which the heart was stopped to mimic donation after cardiac death (DCD), there was a massive increase in apoptosis, caspase-3/7 activity, and caspase-3 protein expression.
189 Apoptosis was compared in DCD kidneys subjected to static versus pulsatile perfusion for 24 hours. Pulsatile perfusion significantly reduced proximal tubular apoptosis and was associated with increased Bcl-2 and hypoxia-inducible transcription factor-1α in the kidney.
The relevance of these studies to organ preservation and the subsequent risk of graft dysfunction is substantial. Caspase inhibitors are a particularly attractive approach to reduce the incidence of DGF in kidney transplantation.
Caspase-1 and IL-18
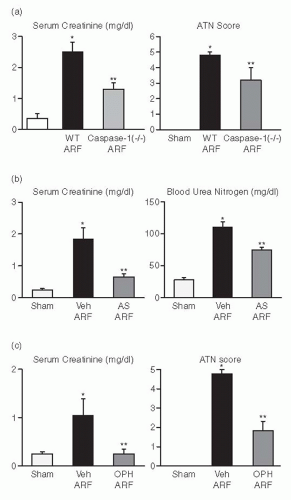
Caspase-1 is a proinflammatory caspase that cleaves precursor IL-1β and precursor IL-18. Caspase-1-/- mice developed less ischemic AKI as judged by renal function and renal histology (
Fig. 29.6A).
190 IL-1β receptor knockout mice or mice treated with IL-1β receptor antagonist (IL-1Ra) are not protected against ischemic AKI.
30 Because caspase-1 also activates IL-18, lack of the mature form of IL-18 in these caspase-1-/- mice was investigated as a possible mechanism of this protection against AKI. Kidney IL-18 was more than 100% increased in wild-type AKI as compared to sham-operated controls. On an immunoblot analysis, there was
a conversion of the precursor to the mature form of IL-18 in AKI wild-type mice, but not in the caspase-1-/- AKI mice and sham-operated controls. To further analyze the role of IL-18, wild-type mice were injected with rabbit antimurine IL-18 neutralizing antiserum prior to the ischemic insult. These mice were protected against AKI to a similar degree as caspase-1-/- mice (
Fig. 29.6B).
Caspase-deficient mice have provided extensive information on the role of individual caspases in disease processes. The study of caspase inhibitors is an important step toward the possible therapeutic effect of caspase inhibition in ischemic AKI. Mice with ischemic AKI treated with newly developed caspase inhibitor, Q-VD-(Ome)-OPH (OPH-001) had a marked reduction (100%) in BUN and serum creatinine and a highly significant reduction in the morphologic AKI score compared with vehicle-treated mice (
Fig. 29.6C).
191 OPH-001 significantly reduced the increase in caspase-1 activity and IL-18 and prevented neutrophil infiltration in the kidney during ischemic AKI. To further investigate whether this lack of neutrophil infiltration was contributing to the protection against ischemic AKI, a model of neutrophil depletion was developed. Neutrophil-depleted mice had a small (18%) reduction in serum creatinine during ischemic AKI but no reduction in the AKI score despite a lack of neutrophil infiltration in the kidney. Remarkably, caspase-1 activity and IL-18 were still significantly increased in the kidney in neutrophil-depleted mice with AKI. Thus, to investigate the role of IL-18 in ischemic AKI in the absence of neutrophils, neutrophil-depleted mice were treated with an IL-18-neutralizing antiserum. IL-18-antiserum-treated neutrophil-depleted mice with ischemic AKI had a significant reduction (75%) in serum creatinine and a significant reduction in the AKI score compared to vehicle-treated neutrophil-depleted mice. These results suggest a novel neutrophil-independent mechanism of IL-18-mediated ischemic AKI.
In other studies, it was determined whether IL-18— binding protein transgenic (IL-18BP Tg) mice are protected against ischemic AKI.
192 IL-18 function is neutralized in IL-18BP Tg mice. IL-18BP Tg mice with AKI had significantly lower BUN, serum creatinine, and ATN score than wild-type mice. The number of macrophages in the kidney was significantly reduced in IL-18BP Tg compared with wild-type mice. The proinflammatory chemokine, CXCL1 (also known as KC or IL-8), was significantly reduced in the kidneys of IL-18BP Tg mice compared to wild-type mice. This study demonstrates that protection against ischemic AKI in IL-18BP Tg mice is associated with less macrophage infiltration and less production of CXCL1 in the kidney.
The effects of different caspase inhibitors on ischemic AKI in the rat kidney have been studied.
193 A caspase-1 inhibitor significantly reduced functional and histologic evidence of ischemic AKI compared to a caspase-3 inhibitor. Another group of investigators found that caspase-1—deficient mice were not protected against renal ischemia.
194 In this study, the model of renal ischemia was 45 minutes of unilateral renal pedicle clamping with contralateral nephrectomy. This model produces a milder form of functional injury than bilateral clamping. At 24 hours, BUN and creatinine were lower in the caspase-1-/- mice than in the wild-type, but the decrease was not statistically significant.
Matrix Metalloproteinases
Matrix metalloproteinases are a large family of zinc-dependent matrix-degrading enzymes that include interstitial collagenases, stromelysins, gelatinases, elastases, as well as membrane-type matrix metalloproteinases. They play a crucial role in remodeling the extracellular matrix, which is an important physiologic feature of normal growth and development. In the kidney, interstitial sclerosis and glomerulosclerosis have been associated with an imbalance of extracellular matrix synthesis and degradation.
195 Alterations in renal tubular basement membrane matrix proteins, laminin and fibronectin, occur after renal ischemia-reperfusion injury.
196 The role of matrix metalloproteinases in this process has been studied.
In endothelial cells isolated from ischemic kidneys, the proteolytic activity of proMMP-2, proMMP-9, and MMP-9 was increased. Occludin, an in vivo MMP-9 substrate, was partly degraded in the endothelial fractions during ischemia, suggesting that the upregulation of MMP-9 had a functional effect to degrade occluding. These data suggest that AKI leads to the degradation of the vascular basement membrane and to increased permeability related to the increase of MMP-9.
197 In renal cells, in vitro cleavage of cadherins in normal rat kidney (NRK) cells requires active membrane-type (MT)1-MMP (MT1-MMP), also known as MMP-14.
198 The disruption of cadherin/catenin complexes in AKI may be associated with the transtubular back-leak of glomerular filtrate. In contrast to the potential injurious role of some MMPs, MMP9 protects the S3 segment of the proximal tubule and the intercalated cells of the collecting duct from apoptosis in AKI, most likely by releasing soluble stem cell factor (sSCF), an MMP9 substrate.
199Meprin A is a zinc-dependent metalloendopeptidase that is present in the brush border membrane of renal proximal tubular epithelial cells. The redistribution of this metalloendopeptidase to the basolateral membrane domain during AKI results in degradation of the extracellular matrix and damage to adjacent peritubular structures. The effect of meprin A, the major matrix degrading metalloproteinase in rat kidney, on the laminin—nidogen complex was examined. Nidogen-1 (entactin) acts as a bridge between the extracellular matrix molecules, laminin-1 and type IV collagen, and thus participates in the assembly of basement membranes. Following ischemic injury, meprin A undergoes redistribution and/or adherence to the tubular basement membrane. Nidogen breakdown products are produced as the result of partial degradation of tubular basement membrane by meprin A following renal tubular ischemia—reperfusion injury.
200The susceptibility of inbred strains of mice to ischemic and nephrotoxic acute renal failure was studied in mice with normal and low meprin A activity.
201 The strains of mice with normal meprin A developed more severe renal functional and structural injury following renal ischemia or the injection of hypertonic glycerol compared to the two low meprin A strains. These findings suggest that meprin A plays a role in the pathophysiology of AKI following ischemic and nephrotoxic acute renal failure insults to the kidney.
201 A recent study
202 demonstrated that meprin inhibition protects against ischemic AKI in vivo in rats.
Nitric Oxide
NO is a lipophilic, highly reactive free radical gas with diverse biomessenger functions.
203 NO mediates diverse functions including vasodilatation, platelet aggregation inhibition, neurotransmission, inflammation, antimicrobial and antitumor actions, and apoptosis.
203 Whether the net effects of NO are beneficial or deleterious is determined by the cell type, the concentration of NO, the duration of production, and the composition of the surrounding microenvironment.
203 There are three major nitric oxide synthase (NOS) isoforms in the kidney: neuronal NOS or nNOS (also known as NOS1), inducible NOS (iNOS, also known as NOS2), and endothelial NOS or eNOS (also known as NOS3) (
Table 29.5).
204 The macula densa is the principal site of nNOS expression in the kidney.
205,
206 In situ hybridization studies in the NRK demonstrate iNOS mRNA in the S3 segment of the proximal tubule, the cortical and medullary thick ascending limb, the distal convoluted tubule, and the cortical collecting duct and the inner medullary collecting duct.
207 eNOS mRNA has been detected in glomeruli, preglomerular vasculature, and proximal and distal tubules.
208 eNOS protein is mainly present in the endothelium of intrarenal, afferent, efferent, and glomerular arterioles and the medullary vas recta.
205 Expression of eNOS protein in tubules has not yet been reported.
204 nNOS and eNOS are continuously present, are activated by calcium, and are also termed constitutive NOS (cNOS).
209,
210 In contrast, iNOS is induced when the cells have been stimulated by certain cytokines, microbes, and microbial products, and thus is called iNOS.
211,
212 The time course of both calcium-dependent and -independent NOS activity in the rat renal cortex and medulla has been studied.
213 Calcium-dependent NOS activity in the cortex and the medulla decreased in the early phase of AKI and then increased in the recovery phase in the cortex. iNOS activity increased in the early phase of AKI in both the cortex and the medulla and was maintained at higher levels in the medulla. However, in another study, L-arginine improved the deficiency of constitutive NOS activity and improved the recovery phase of ischemic AKI in rats.
214Studies in freshly isolated proximal tubules from knockout mice have also revealed the role of NO in hypoxic/ischemic tubular injury. Hypoxia-induced proximal tubule damage, as assessed by LDH release, was no different between wild-type and mice in which eNOS and nNOS had been knocked out. However, proximal tubules from iNOS knockout mice demonstrated resistance to the same degree of hypoxia.
42 The iNOS knockout mice also had less renal failure and better survival than the wild-type mice after renal artery clamping.
215 An induction of heat shock protein was also observed in the iNOS knockout mice as a potential contributor
to the protection. Chiao et al.
216 produced further results in a renal artery clamp model in mice in which alpha melanocyte-stimulating hormone (αMSH) was shown to block the induction of iNOS, decrease neutrophil infiltration, and afford functional protection. A subsequent study examined the relative importance of αMSH on the neutrophil pathway by examining the effects of αMSH in ICAM-1 knockout mice and neutrophil-poor isolated perfused kidneys where neutrophil effects are minimal or absent.
217,
218 In this study, it was found that αMSH decreases renal injury when neutrophil effects are minimal or absent, indicating that αMSH inhibits neutrophil-independent pathways of renal injury.
Hypoxia was found to increase NO release from freshly isolated proximal tubules and this effect was blocked by L-NAME, a nonspecific NOS inhibitor, but not by the inactive D-NAME compound.
39,
219 The NO release during hypoxia was accompanied by LDH release and was reversed by L-NAME administration. Interestingly, however, the administration of L-NAME, the nonspecific NOS inhibitor, to the rat kidney clamp model actually worsened the renal failure.
220 This result was interpreted as an overriding blocking effect of eNOS activity with the nonspecific effects of L-NAME.
68 This would worsen the renal vasoconstriction and the resultant injury, thus obscuring any salutary effect at the level of the proximal tubule.
221 Thus, opposing abnormalities in NO production within the endothelial and tubular compartments of the kidney may contribute to renal injury.
68 Reduced eNOS-derived NO production causes vasoconstriction and worsens ischemia; increased iNOS-derived NO production by tubular cells adds to the injurious effects of ischemia on these cells. Therapeutic interventions to modulate NO production in ischemic AKI may require the selective modulation of different NOS isoforms in the tubular and vascular compartments of the kidney.
222 On this background, Noiri and colleagues
220 performed studies using a specific antisense oligonucleotide to iNOS. The ischemia-induced upregulation of iNOS and nitrite production were both blocked by the antisense oligonucleotide. Most importantly, the BUN and serum creatinine did not rise after the renal ischemic insult in the animals treated with the antisense oligonucleotide against iNOS.
Noiri and colleagues also studied the relationship between NO and osteopontin during ischemic AKI. Osteopontin is a negatively charged glycosylated phosphoprotein that is expressed in many tissues, including the renal epithelial cells. Osteopontin serves both a cell attachment function and a cell signaling function via the alpha-v beta-3 integrin. Effects on gene expression include suppression of the induction of NOS by inflammatory mediators. Osteopontin may play an important role in the pathophysiology of AKI. Osteopontin knockout mice subjected to renal ischemia developed worse renal failure and more structural damage than wild-type controls.
223 This was associated with the augmented expression of inducible NOS and the prevalence of nitrotyrosine residues in kidneys from osteopontin knockout mice versus wild-type counterparts. This study
220 provides strong evidence of the renoprotective action of osteopontin in acute renal ischemia.
The protective effect of 17 β-estradiol against ischemic AKI in rats is due to the activation of the PI3K/Akt pathway followed by increased eNOS phosphorylation.
224The microvillar actin and cellular integrins are potential substrates of NO action, which could contribute to the ischemia-mediated sloughing of the brush border membrane and the detachment of the proximal tubule epithelial cells from their extracellular matrix.
11,
225,
226,
227 Such an effect would not only result in impaired tubular sodium reabsorption, but would also provide intraluminal cellular debris as a component of tubular cast formation.
Heat Shock Proteins
The stress response is a highly conserved homeostatic mechanism that allows cells to survive a variety of different stresses.
228 Stresses that trigger the heat shock response include hyperthermia, hypothermia, the generation of oxygen radicals, hypoxia/ischemia, and toxins.
229 On a molecular level, their function is to protect cells from environmental stress damage by binding to partially denatured proteins, dissociating protein aggregates, regulating the correct folding, and cooperating in transporting newly synthesized polypeptides to the target organelles.
The proteins induced by these stresses belong to a family of proteins called heat shock proteins (HSP). The proteins are identified by their molecular weight. The most important families include proteins of 90, 70, 60, and 27 kDa.
229 HSP 90 is essential for cell viability. It is associated with the steroid hormone receptor and is a general chaperone with ATPaselike activity. In stressed cells, it associates with the cytoskeletal protein, actin. The HSP 70 family includes proteins that are both constitutively expressed and induced by stress. They are the most highly induced proteins by stress and function as chaperones, binding to unfolded or misfolded proteins. The HSP family is restricted to the mitochondrial matrix where it functions as an unfoldase. The HSP 27 family has functions similar to HSP 70. Ubiquitin is a stress protein that binds denatured proteins and targets them for proteolysis by the proteasome.
Renal ischemia results in both a profound fall in cellular ATP and a rapid induction of the 70 kD heat shock protein family, HSP-70.
230,
231 The relationship between cellular ATP and the induction of the stress response in the renal cortex during renal ischemia has been studied. Van Why et al.
232 demonstrated that a 50% reduction in cellular ATP in the renal cortex must occur before the stress response is detectable, that a reduction of ATP below 25% of control levels produces a more vigorous response, and that reperfusion is not required for the initiation of a heat shock response in the kidney.
232Ischemic AKI also induces differential expression of small HSPs. In sham-operated kidneys, HSP 25 localized to glomeruli, vessels, and collecting ducts, whereas another
stress protein, αB-crystallin, localized primarily in medullary thin limbs and collecting ducts. After ischemia, HSP 25 accumulated in proximal tubules in the cortex and the outer medulla, whereas αB-crystallin labeling became nonhomogeneous in the outer medulla, and increased in the Bowman capsule. This study demonstrates that there is striking differential expression of HSP 25 and αB-crystallin in various renal compartments.
233In vitro studies have demonstrated that HSP induction protects cultured renal epithelial cells from injury. It has been determined that prior heat stress protects opossum kidney (OK) cells, a cultured renal epithelial cell line, from injury mediated by ATP depletion.
234 Also, HSP 70 overexpression is sufficient to protect cultured proximal tubule (LLC-PK1) cells from hyperthermia but is not sufficient to protect against hypoxia.
235