Exon
Mutation
Mutation type
Ref.
Exon
Mutation
Mutation type
Ref.
1
Del exon 1–2 (19353 bp)
Truncation
[37]
10
1397-1398delTC
Frameshift
Del exon 1–2 (5761 bp)
Truncation
[37]
1466insC
Frameshift
[42]
Del 5’UTR-exon 1 (150 bp)
Truncation
[37]
1470-1483del
Truncation
[43]b
2T > C
Initiation codon
[44]
1472insA
Frameshift
[21]
3G > C
Initiation codon
[45]
1476delAG
Frameshift
[20]
41delT
Frameshift
[46]
1488-1494del CGAGGAC
Frameshift
[15]
45insT
Frameshift
[21]
1507C > T
Nonsense
[47]
46insTGC
Frameshift
[44]
1565 + 1G > A
Splice site
[48]
49-2A >C
Splice site
[41]
1565 + 1G >T
Splice site
[22]
49-2A > G
Splice site
[19]
1565 + 2insT
Splice site
[49]
49G > T
Splice site
[50]
1565 + 2dup
Splice site
[43]
2
53delC
Frameshift
[22]
11
1588insC
Frameshift
[15]
59G > A
Nonsense
[19]
1595G >A
Nonsense
[43]b
70G > T
Nonsense
[15]
1610delC
Frameshift
[51]
3
164-?387+?del
Nonsense
[38]
1619insG
Frameshift
[52]
185G > T
Missense
[53]
1682insA
Frameshift
[24]
187C > T
Nonsense
1679C > G
Missense
[43]
190C > T
Nonsense
[15]
1710delT
Frameshift
[22]
283C > T
Nonsense
1711insG
Frameshift
[17]
353C >G
Missense
[41]
1711 + 5G >A
Splice site
[20]
372delC
Frameshift
[16]
12
1774G >A
Missense
[24]a
377delC
Frameshift
[24]
1779insC
Frameshift
[20]
382delC
Frameshift
[20]
1792C > T
Nonsense
4
469delG
Frameshift
[43]
1795A > T
Missense
[24]a
531 + 2T > A
Splice site
[55]
1849G > A
Missense
[56]
5
586G > T
Nonsense
[15]
1901C > T
Missense
687 + 1G > A
Splice site
[20]
1913G > A
Nonsense
[24]
6
715G > A
Missense
13
2061delTG
Frameshift
[20]
731A > G
Missense
2064delTG
Frameshift
753insG
Frameshift
[44]
2095C >T
Nonsense
7
Del exon 7–16
Truncation
[39]
2161C > G
Splice site
[45]
832G > A
Splice site
2164 + 2T > A
Splice site
[43]
832 + 1G > T
Splice site
[43]
2164 + 5G > A
Splice site
833-2A > G
Splice site
14
Del exon 14–16 (8078 bp)
Truncation
[37]
892G > A
Missense
[20]
2195G > A
Missense
1003C > T
Nonsense
2245C > T
Missense
[24]
1008G > T
Splice site
[12]
2269G > A
Missense
[63]
8
1017delC
Frameshift
[41]
2275G > T
Nonsense
[64]
1018A > G
Missense
2276delG
Frameshift
[45]
1023T >G
Nonsense
[44]
15
2295 + 5 G > A
Splice site
[22]
1062delG
Frameshift
[24]a
2287G > T
Nonsense
[66]
1064insT
Frameshift
[20]
2310delC
Frameshift
[20]
1107delC
Nonsense
[41]
2329G > A
Missense
[24]
1118C > T
Missense
[67]
2343A > T
Missense
1134del8ins5
Frameshift
[20]
2381insC
Frameshift
[12]
1135 + 5del8ins5
Splice site
[68]
2386delC
Frameshift
[43]
1137G > A
Splice site
2395delC
Frameshift
[49]
1137 + 1G > A
Splice site
[15]
2396C > G
Missense
[52]
9
1147C > T
Nonsense
[43]
2398delC
Frameshift
1189A > T
Missense
[69]
2399delG
Frameshift
[70]
1212delC
Frameshift
2440-1C > T
Splice site
[71]
1225T > C
Missense
[20]
2440-6C >G
Splice site
[41]
1285C > T
Missense
[45]
16
Del exon 16 (828 bp)
Truncation
[37]
1306_1303insA, 1306_1307delTT
Frameshift
[72]
As per Knudson’s two-hit hypothesis of tumour suppressor gene inactivation, a second event is required to account for loss or inactivation of the wild-type CDH1 allele [73]. Promoter hypermethylation of the second CDH1 allele has been demonstrated by several groups as the most common mechanism inactivating the wild-type CDH1 allele in HDGC [42, 74]. CDH1 promoter hypermethylation has been found in prostate, breast and sporadic GC [75, 76], and was demonstrated by Grady et al. in 2000 to be the “2nd hit” in some HDGC patients [77]. Grady et al. also demonstrated in vitro that the demethylating agent, 5-azacytidine restored E-cadherin expression in a GC cell line that tested positive for CDH1 promoter methylation, revealing that methylation was the mechanism of silencing. Other mechanisms include somatic mutation, one case of an intragenic deletion has been identified, and it is thought that histone modifications may also be important [42, 68, 74, 78]. In GC tumours from HDGC patients’, loss of heterozygosity is another mechanism for loss of the wild-type CDH1 allele [12, 15, 19, 74].
It is not uncommon for HDGC patients to have multiple foci of tumour in their gastrectomy specimens. Genetic analysis of multiple tumours in the same individual reveal that different mechanisms of silencing of the second allele occur independently at multiple sites in metastatic deposits [74] and within lesions of the stomach [79] .
Non-CDH1 Hereditary Diffuse Gastric Cancer
About 25 to 30 % of patients meeting current clinical criteria for HDGC are found to have germline mutations in CDH1, meaning that up to 70 % of families have no identifiable mutation [7]. Using the nomenclature reported by Blair et al., families that fulfil the IGCLC criteria for HDGC (see below) but have no identified CDH1 mutation are designated familial diffuse gastric cancer (FDGC) , HDGC refers only to families with a pathogenic CDH1 mutation [10]. CDH1 genetic testing involves sequencing and multiplex ligation-dependent probe amplification (MLPA) to detect large deletions. While this testing is currently the gold standard, there may still be mutations that are missed due to technological limitations. Families without identified CDH1 mutations have been investigated for other potential candidate genes involved in FDGC. In a Dutch kindred with FDGC, a mutation has recently been identified in CTNNA1, which encodes alpha-E-catenin, making this a potential causative mutation in FDGC [80]. Alpha-E-catenin, in a complex with beta-catenin, binds the cytoplasmic domain of E-cadherin to the cytoskeleton [32, 81, 82]. Loss of CTNNA1 in animal models induces altered cell polarity, hyperproliferation, and increase in Ras- and mitogen-activated kinase (MAPK) activity—features which are consistent with the potential to induce a malignant phenotype [83]. While the CTNNA1 mutation in this family is suspicious of pathogenicity for HDGC, the phenotype suggests older onset of DGC and mutations in CTNNA1 have not been found in other families to validate the result. It is likely that some families with FDGC may harbour mutations in other genes that have yet to be identified.
Other Malignancies Associated with HDGC
Other malignancies have shown higher prevalence in families with HDGC, the most prominent of which is LBC [10, 16, 18, 48]. The risk for developing LBC for females with CDH1 mutations is approximately 60 % by age 80 [7]. Therefore, LBC is considered a cancer in the HDGC syndrome that warrants specific management. Colorectal cancer has been identified in some HDGC kindreds [19, 20, 41], although the numbers are small and given this is such a common cancer in the community, direct pathogenesis from a CDH1 mutation has not been established [18].
Other Hereditary Syndromes Associated with Gastric Cancer
Other hereditary syndromes are associated with increased risk for GC, including Lynch syndrome or hereditary non-polyposis colorectal cancer (HNPCC) , hereditary breast and ovarian cancer, familial adenomatous polyposis (FAP), Cowdens syndrome and Peutz–Jeghers syndrome. Table 6.2 describes the associated risk of GC , which can be intestinal or diffuse type, with selected syndromes where it is known. Patients with hereditary breast and ovarian cancer syndrome have increased risks of malignancies in addition to breast and ovarian cancer, including gastric cancer although the risks are not well quantified [84, 85]. Patients with Lynch syndrome, in addition to colorectal cancer, are at risk of GC, other gastrointestinal malignancies, endometrial carcinoma and transitional cell carcinomas of the renal tract [6, 86, 87].
Table 6.2
Inherited cancer syndromes with associated GC risk
Cancer syndrome | Gene | Gastric cancer risk lifetime risk (%) | Ref. |
|---|---|---|---|
HDGC | CDH1 | 80 | [7] |
Hereditary breast/ovarian cancer | BRCA1 | 5.5 (3.4–7.5) | [85] |
BRCA2 | 2.6 (1.5–4.6) | [84] | |
Lynch syndrome | MLH1, MSH2, MSH6, PMS2 | 4.4–19.3 | |
Peutz–Jeghers syndrome | STKII | 29 | [88] |
Polyposis of the gastrointestinal (GI) tract is associated with a number of familial cancer syndromes. Upper GI polyposis is frequently found associated with FAP. In the stomach this is manifested as fundic gland polyposis [89, 90]. Although sporadic fundic gland polyps are considered to be non-neoplastic, in FAP patients some have been found to harbour dysplasia which may evolve into invasive GC [89, 91, 92]. Peutz–Jeghers and Cowden syndrome patients present with polyposis of multiple organs. Only rare cases of GC have been reported in Cowden syndrome, suggesting this is not a common manifestation of this disease [93, 94]. The risk of cancer in Peutz–Jeghers syndrome is higher, with a life-time risk of GC of 29 % [88]. Juvenile polyposis syndrome is another syndrome associated with hamartomatous polyp formation of the GI tract, with associated increased risk of GI malignancy mainly related to colorectal cancer, but GC has also been documented [95, 96]. GC has also been seen in families with Li–Fraumeni syndrome [97, 98], and some FDGC families have been identified with TP53 mutations suggesting GC is a component of Li–Fraumeni syndrome spectrum [57]. Although there appears to be no significant increase in the incidence of GC in patients with MUTYH-associated polyposis, a autosomal recessive disorder caused by germline mutations in the base excision repair gene MUTYH [99], monoallelic MUTYH mutation carriers have been reported to have a higher incidence of GC than the general population [100]. There are reports of an increased incidence of GC in relatives of patients with Fanconi’s anaemia [101]. In 2012 a new autosomal dominant condition associated with gastric polyposis and intestinal GC was described. This featured proximal polyposis of the stomach and GC and was named gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS) [102, 103]. The causative gene for this syndrome is unknown.
Pathology
Mechanism of Tumorigenesis
E-cadherin is involved in cell adhesion, epithelial-to-mesenchymal transition (EMT) and in regulation of Wnt signalling via beta-catenin. Although the exact mechanisms by which loss of E-cadherin instigates tumorigenesis remain to be elucidated, it is thought that in HDGC, CDH1 acts as a tumour suppressor gene, with loss of function leading to loss of cell adhesion with subsequent invasion and metastasis. One hypothesis is that loss of E-cadherin and subsequent loss of cell adhesion causes disruption to cell polarity that interferes with cell division and results in daughter cells being deposited in the lamina propria, which then expand forming foci of SRCs [78]. The in vitro and in vivo evidence available suggests that the loss of cell adhesion alone caused by deficient E-cadherin expression is sufficient to initiate DGC.
Loss of E-cadherin expression is one of the hallmarks of EMT [104]. EMT is the process by which epithelial cells assume a more mesenchymal phenotype, including the ability to migrate through the basement membrane and possess some resistance to apoptosis [105]. This process is normal in human development, but is pathological when implicated in tumour progression and metastasis. In addition to disrupting cell adhesion and therefore potentiating an invasive phenotype, loss of E-cadherin expression also promotes dysregulated beta-catenin signalling through the canonical Wnt signalling pathway which has been associated with tumorigenesis in a wide variety of cancers [106].
E-cadherin ’s role as a suppressor of tumour invasion has been shown in vitro, where loss of expression or function leads to altered cell phenotype and enhanced cell invasiveness [27]. This phenotype can be reverted by restoring E-cadherin protein expression after transfection of E-cadherin coding cDNA [107, 108].
The important role that E-cadherin has in embryogenesis is reflected in the fact that E-cadherin homozygous knockout mice are embryonic lethal [109]. Heterozygous mutant animals are phenotypically normal, and have been used to establish a mouse model of DGC by exposure to a carcinogen (N-methyl-N-nitrosurea) to induce the 2nd hit [79, 109]. It was noted that compared to wild-type treated mice, Cdh1 +/− mice developed intramucosal SRCCs 11 times more frequently. In addition to loss of E-cadherin expression, the SRCCs showed a low proliferative activity and absence of nuclear beta catenin accumulation, suggesting that in the absence of increased proliferation or Wnt signalling activation, loss of cell-to-cell adhesion alone was sufficient to initiate a DGC in these models [79]. This is consistent with the clinical observation, where hundreds of independent foci of SRCCs can occur in the stomachs of patients with germline CDH1 mutations, which suggests that it is unlikely that other genes are required to initiate HDGC [78].
It is not known why germline CDH1 mutations predispose to DGC, and LBC, but not significantly to other malignancies. One hypothesis is the higher carcinogen exposure and chronic inflammation that the gastric epithelium is exposed to, another is the high cellular turnover of the gastric epithelium [78]. In these settings, fewer mutational or epigenetic events may be required to generate an invasive malignant phenotype. It is notable, however, that epithelial cell turnover in the intestine is also high, and why colorectal cancer is not more apparent in patients with CDH1 mutations remains unresolved .
Risk Modifiers
There has not been a comprehensive analysis of the genetic or environmental factors that impact on penetrance of HDGC. Knowledge of these would be beneficial for genetic counselling on risk behaviours and on genetic risk profile. However, there are a number of lifestyle and environmental factors that have been identified to impact on risk of sporadic GC (see Chaps. 1–3), and it is possible that these factors also have impact on HDGC.
Helicobacter Pylori
Helicobacter pylori (H. pylori) was classified as a class I carcinogen by the World Health Organisation in 1994, and has a well-established association with GC [110]. It has been implicated in both sporadic intestinal GC, and DGC, and although it does not appear to be required for oncogenesis in HDGC [66, 111–113], it theoretically may modulate disease risk [114]. Current recommendations for HDGC suggest eradicating H. pylori if found [10], but there are no prospective analyses showing impact of this on progression of HDGC.
Physical Activity and Diet
Numerous studies have investigated the association of sporadic GC with demographics, diet and physical activity; however, there are no studies that investigate HDGC specifically or as a subgroup.
Diet and food storage have been implicated in the changing incidence of sporadic GC, with the adoption of refrigeration improving access to fresh fruit and vegetables and reducing the need for salt preservation of food being implicated in the decrease in incidence observed in GC [115–118]. The European Prospective Investigation into Cancer and Nutrition (EPIC) study was designed to prospectively investigate relationships between cancer incidence and lifestyle, genetic and environmental factors. It found an inverse association with vegetable intake for intestinal GC, but not diffuse GC, and no association with fruit intake [119]. For sporadic GC, the EPIC study demonstrated an inverse association with physical activity, particularly in non-cardiac GC [120]. Findings of the EPIC study also showed an association between smoking and sporadic GC, with approximately 18 % of cases in this study being attributable to smoking [121]. Whether smoking, physical activity, diet or other environmental factors impact on the penetrance of HDGC is unknown, but would be assumed to have similar affects as in sporadic GC.
CDH1 Mutations in Sporadic GC
E-cadherin mutations were identified in sporadic GC prior to the identification of CDH1 as the gene responsible for HDGC [29]. Inactivating somatic mutations in CDH1 are detected in over 50 % of sporadic DGC, but not intestinal GC [29, 122]. As with HDGC, promoter methylation of CDH1 has been identified in sporadic DGC as the 2nd hit [122]. The high prevalence of mutation and epigenetic silencing of CDH1 suggests that the inactivation of E-cadherin has a role in the evolution of sporadic DGC as well as HDGC, with the earlier age of onset of GC seen in HDGC kindreds reflecting the fact that only one wild-type CDH1 allele requires mutation to develop a potentially malignant genotype. Interestingly, although mutations in CDH1 are not seen in intestinal GC, promoter methylation has been identified in a subset of intestinal GC [76, 122]. It is likely that silencing of E-cadherin expression has an effect on the later stages of the carcinogenic cascade in intestinal type GCs, whereas it has a role in early pathogenesis of DGC .
Management of HDGC
Clinical Criteria for Genetic Testing
Figure 6.1 describes the suggested process for the diagnosis and management of HDGC. The most recent published guidelines for determining which individuals to test for CDH1 mutations from the IGCLC were published in 2010 [7]. They recommend genetic testing for patients who meet the following criteria:
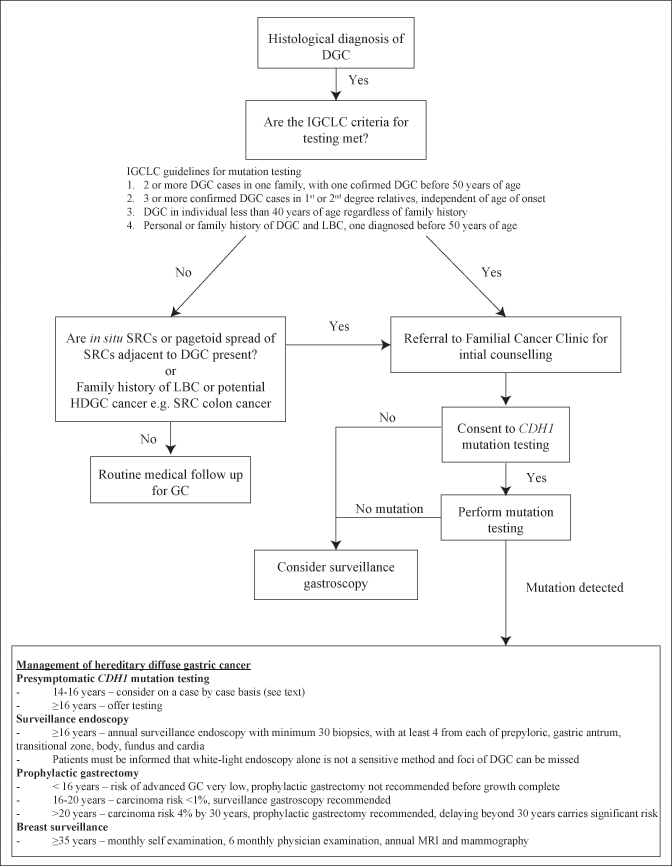
Two or more GC cases in one family, with one confirmed DGC before 50 years of age
Three or more confirmed DGC cases in 1st or 2nd degree relatives, independent of age of onset
DGC in individual less than 40 years of age regardless of family history
Personal or family history of DGC and LBC, one diagnosed before 50 years of age
It is expected that these guidelines will be updated in 2014 and may provide changes in the inclusion criteria. In addition to the above guidelines, the IGCLC recommends consideration to offering genetic testing to patients where pathologists identify in situ SRCs or pagetoid spread of SRCs adjacent to DGC, as this is rarely seen in sporadic DGC [123].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:
 Epidemiology of Gastric Cancer
Epidemiology of Gastric Cancer
 Lymphadenectomy—D1, D2, and D3
Radiation Treatment for Gastric Cancer
Lymphadenectomy—D1, D2, and D3
Radiation Treatment for Gastric Cancer
 Methods of Reconstruction—BI, BII, Roux-en-Y, Jejunal Interposition, Proximal Gastrectomy and Pouch Reconstruction
Methods of Reconstruction—BI, BII, Roux-en-Y, Jejunal Interposition, Proximal Gastrectomy and Pouch Reconstruction
 Endoscopy and Endoscopic Ultrasound Examination of the Stomach
Endoscopy and Endoscopic Ultrasound Examination of the Stomach
 Pathology of Gastric Cancer
Pathology of Gastric Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


