Fig. 2.1
Gastric cancer (GC) histotypes according to Laurèn classification. a. Intestinal-type GC: glandular cancer structures of different size infiltrating the gastric wall (H&E, original magnification 40 ×). b Diffuse-type GC: noncohesive cancer cells infiltrating the gastric wall, without forming glandular structures (H&E, original magnification 20 ×)
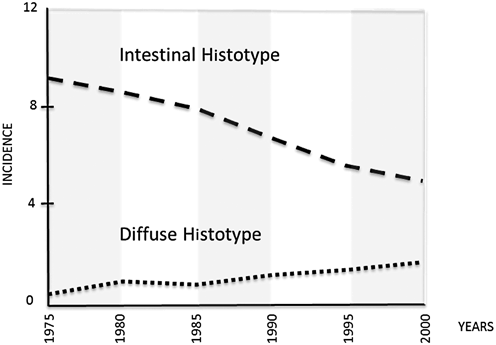
Fig. 2.2
In the USA, the incidence of the GC intestinal histotype progressively decreased between 1975 and 2000; during the same time interval, an increasing incidence of diffuse histotype was noted. (From Arch Pathol Lab Med. 2004; 128: 765–70; modified)
Other widely used classifications include that of the World Health Organization (WHO) [9] and the one proposed by the Japanese Gastric Cancer Association [22].
Etiology
Gastric cancer is a multifactorial disease. It can be syndromic/hereditary, associated with specific mutational profiles [23–26]. Most frequently, however, gastric cancers are sporadic and stem from a progressive accumulation of genotypic and phenotypic changes triggered by longstanding gastritis, primarily due to H. pylori infection [27–29].
In 1994, the International Agency for Research on Cancer (IARC) recognized H. pylori infection as a type I carcinogen [30]. It has been estimated that H. pylori infection is responsible for more than 75 % of distal (antral) gastric cancers and associated with both types, intestinal and diffuse; its association with proximal (cardia-based) carcinomas is more dubious [31].
H. pylori is a Gram-negative spiral bacterium with a variety of mechanisms that enable it to colonize the gastric mucosa and evade or modify the host’s immune response [32]. The infection is usually acquired in childhood and typically persists for decades unless it is treated and the bacterium is eradicated. The exact mechanisms behind the bacterium’s transmission are still unknown, but it is believed to be transmitted person to person.
Multiple mechanisms have been described for the carcinogenesis associated with H. pylori, including inflammation, direct interactions with organisms that cause genetic instability in the host, and H. pylori-associated epigenetic alterations [33]. H. pylori differ in terms of their virulence. The most important factors influencing H. pylori virulence include a vacuolating toxin, VacA, H. pylori neutrophil-activating protein (NapA), and proteins encoded by the cag pathogenicity island [34].
H. pylori is believed to have a necessary, but not sufficient causative role in gastric cancer. For instance, the lifetime risk of gastric cancer in the Japanese has been estimated at 11 %, whereas gastric cancer is rare among South Africans or Southern Indians, despite a very high prevalence of H. pylori infection [35]. As discussed below, H. pylori-associated gastric cancer is closely associated with atrophic gastritis, which is associated in turn with environmental factors, and especially diet and gastric cancer incidence changes rapidly with migration or diet.
Epstein-Barr virus (EBV) is another infectious agent involved in gastric cancer, and several Asian, European, and American studies have consistently associated EBV infection with 5–16 % of gastric cancers [36, 37]. Male patients were twice as likely to have EBV-positive tumors as female patients, and tumors arising in the proximal stomach were more than twice as likely to be EBV-positive as tumors in the antrum. No difference in the prevalence of EBV has been demonstrated between the intestinal and diffuse histological types, but a strong association (> 90 %) has been established between EBV infection and the uncommon lymphoepithelioma-like gastric cancer. The role of EBV in gastric carcinogenesis is, however, yet to be clarified.
Noninfectious Environmental Factors and Lifestyle Variables
Among the dietary factors, high salt intake has historically been associated with a higher risk of gastric cancer, mainly in association with H. pylori infection [8]. Many case-control studies consistently demonstrated a positive association between salted fish/meat, salted vegetables, and gastric cancer, and this association was recently confirmed in a systematic review of the available epidemiological data [38, 39].
A diet rich in meat has also been suggested as a risk factor in Europe. A large-scale European study found a significant correlation between meat consumption and distal gastric cancer, and this association was stronger in subjects infected with H. pylori [40].
Tobacco smoking is a risk factor for the onset of gastritis, ulcers, and both proximal and distal gastric cancers. It has been claimed that tobacco has an etiological role in up to 18 % of gastric cancer cases, and there is evidence to support the interaction between tobacco smoking and H. pylori infection [41]. In European males and females, both, the intensity and the duration of smoking habits have been associated with the risk of gastric cancer, and proximal cancer in particular (HR = 4.10) [41].
The available information on the etiological role of alcohol consumption is contradictory. A strong association was demonstrated by a Russian case-control study identifying a three-fold higher risk of cardia cancer among male heavy drinkers (OR 3.4; CI 1.2–10.2) [42]. On the other hand, a recent meta-analysis of 44 case control and 15 cohort studies (covering 34,500 cases of gastric cancer) showed that a light/moderate alcohol consumption coincided with an insignificant increase in this risk [43]. A low total body iron, in terms of serum ferritin levels, has also been associated with gastric cancer, but this is probably a spurious link since H. pylori is associated with iron deficiency (the virus would delete iron) [44].
Fruit and vegetables have consistently been attributed a protective role. In a prospective study on 70,000 subjects (139 with gastric cancer), a daily intake of 2–5 servings of fruit/vegetables resulted in a hazard ratio of 0.56 (95 % CI: 0.34–0.93) when compared with less than one serving a day [45].
Host Factors
The risk of gastric cancer has been associated with numerous genetic polymorphisms, mainly involving inflammation-related genes (e.g., IL1B, IL1RN, IL10, and TNF) [8]. Both, interleukin (IL)-1β and tumor necrosis factor (TNF)-α, are powerful proinflammatory cytokines that also suppress gastric acid production. IL-10 is an anti-inflammatory cytokine that counteracts the effects of proinflammatory cytokines, and variants of IL10 have been identified that influence its production. Generally speaking, more virulent H. pylori strains and gene polymorphisms associated with an enhanced inflammatory reaction carry a greater risk.
The risk of developing gastric cancer is 2–10 times higher in subjects with a family history of gastric cancer [40]. Most familial cases are considered sporadic, however, and seem to be influenced by shared environmental factors, such as H. pylori infection, diet, and socioeconomic status. Gastric cancer can nonetheless develop as part of a familial cancer syndrome, such as hereditary diffuse gastric cancer syndrome, Lynch syndrome, familial adenomatous polyposis, Peutz-Jeghers syndrome, and Li-Fraumeni syndrome [49].
Hereditary, diffuse-type gastric cancer is a rare, autosomal dominant disorder responsible for 1–3 % of familial cases. This syndromic cancer is caused by various mutations of the gene encoding E-cadherin (CDH1), a cell adhesion protein essential to maintaining the epithelial tissue architecture [50]. These mutations result in a 70–80 % lifetime risk of the onset of gastric cancer; hence, preventive gastrectomy has been suggested. Other conditions that raise the risk of gastric cancer are pernicious anemia, Menetrier’s disease, and gastric stumps after gastric surgery.
Epidemiology
The global distribution of gastric cancer differs markedly from that of most other adult tumors. Like the majority of “environmental” cancers, the risk of gastric adenocarcinoma is very low in young age and gradually increases with age, reaching a plateau between 55 and 80 years (depending on the variable interaction of different risk factors) [9]. In general, gastric cancer rates are twice as high in men as in women.
The highest incidence rates in males are found in Eastern Asia (Korea, Mongolia, Japan, and China, with rates between 40 and 60 per 100,000 population), Eastern Europe (around 35 per 100,000), and some Latin American countries, especially Central America and the Andean Region, with rates between 20 and 30 per 100,000 population [8, 51]. Some of the lowest incidence rates are found in African countries (0.6–3/100,000) and more recently in North America (5–6/100,000) [8].
It is noteworthy that there are significant differences in the gastric cancer risk for different ethnic groups within the same geographical area [8, 51]. In the USA for example, Hispanics, African-Americans, and Native Indians are more affected than Caucasians [8]. These variations cannot be considered simply as ethnicity-related, however, due to the overlapping disparities in the socioeconomic status, which is also inversely related to both H. pylori prevalence and gastric cancer risk. Several studies conducted in different regions have consistently demonstrated that a low socioeconomic status is per se an adverse variable that raises the risk of (gastric) malignancy [52, 53].
The relevant impact of the “environmental etiological component” is further demonstrated by the lower gastric cancer risk described in the offspring of populations that migrate from high- to low-incidence continents (e.g., from Asia to North America) [54, 55].
In the past 50 years, the incidence rates of stomach cancer have been declining steadily in many parts of the world [27]. This is believed to be partly due to factors associated with the use of refrigerated foods, the availability of fresh fruits and vegetables, and a decrease in the use of salt (at the table and for food preservation). Other likely associated factors include a decrease in the prevalence of H. pylori infection in many countries and the decline in smoking in some industrialized countries [4].
Changing Trends in Incidence by Site
Albeit with some notable exceptions, the incidence of gastric cancer has fallen steadily all over the industrialized world [56]. Looking closer at these epidemiological trends, however, and distinguishing gastric malignancies by their topography, it has been consistently observed that gastric cancers have been “climbing” from the distal to the proximal stomach (Fig. 2.3) [57].
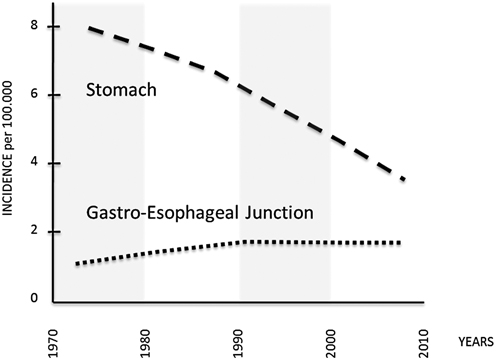
Fig. 2.3
Incidence of adenocarcinoma of the noncardia stomach, and gastroesophageal junction in the USA, 1973–2008 (per 100,000, adjusted for age, race, and sex to the 2000 US standard population, with Lowess smoothing; Data from the National Cancer Institute’s SEER Program). (From Semin Radiat Oncol. 2013;23:3–9; modified)
This phenomenon was initially attributed to an inconsistent recording of tumor topography, but it has been confirmed in many countries, and available data from the Surveillance, Epidemiology, and End Results (SEER) cancer registry program in the USA show an approximate 2.5-fold increase in the incidence of adenocarcinoma at the gastroesophageal junction from 1973 to 1992, with rates stabilizing in the past two decades. Similar proportional trends have been seen among subgroups stratified by race and gender, with significantly higher rates in white males [58–60].
Another phenomenon recently identified in the USA is an increase in the rate of distal gastric cancers in Caucasian adults of both sexes between 25 and 39 years old [8]. This rising trend has continued over the last three decades and its causes are still unknown [56]. In our experience (DYG), these patients are typically young recent immigrants from high incidence areas of Central and South America.
The Genetic Landscape of Gastric Cancer
The molecular profile of gastric cancer is heterogeneous, partly due to different classification systems being used, and also because most analyses have considered a very limited number of cases [50, 61]. As a result, despite the huge amount of data collected, no reliable, novel molecular markers have been introduced for use in secondary prevention strategies to date [50, 62, 63]. Efforts in this direction have gained strength, however, from the recent promising arrival on the scene of innovative technologies (next-generation sequencing [NGS], high-throughput H. pylori microarray know-how) and the unexpected discovery of new classes of biomarkers (microRNA [miRNA], and long noncoding RNAs) [64, 65].
Whole genome sequencing studies have recently revealed new molecules and mechanisms involved in gastric cancers [23–25]. In particular, RHOA mutations have been identified as one of the most important drivers of diffuse-type, but not intestinal-type tumors [24, 25]. Data on 295 primary gastric cancers involved in The Cancer Genome Atlas (TCGA) project point to a new four-tiered molecular classification of gastric cancers : (i) gastric cancers positive for Epstein-Barr virus , which display recurrent PIK3CA mutations, extreme DNA hypermethylation, and amplification of JAK2, PD–L1 and PD–L2; (ii) microsatellite unstable gastric cancers, which show elevated mutation rates; (iii) genomically stable gastric cancers, which are enriched for the diffuse histological variant and mutations of RHOA or fusions involving RHO-family GTPase-activating proteins; and (iv) gastric cancers with chromosomal instability, which show marked aneuploidy and focal amplification of receptor tyrosine kinases [23].
Whole exome sequencing studies have further dissected gastric intestinal-type carcinogenesis [66, 67]. The genes found frequently mutated included TP53, PIK3CA, FAT4, and ARID1A [66, 67]. The latter two have been identified as novel gastric cancer markers. FAT4 is a member of the cadherin gene family, which is mutated in 5 % and deleted in 4 % of gastric cancers [67]. The protein encoded by ARID1A is an accessory subunit of the SWI-SNF chromatin remodeling complex involved in processes of DNA repair, differentiation and development, as well as in the homeostasis of cell proliferation [68]. Mutations in chromatin remodeling genes, such as ARID1A, MLL3, and MLL, have been found in 47 % of gastric cancers [7].
Alterations of the TP53 gene are associated throughout the spectrum of histological lesions involved in gastric oncogenesis. Loss of heterozygosity at the TP53 locus has been demonstrated in 14 % of a series of gastric IM and in 22 % of dysplastic lesions [69]. In a recent series of 15 matched high-grade IEN and early gastric cancers, NGS analysis of hotspot regions in 50 cancer-associated genes disclosed a molecular similarity between the two lesions, and further supported a relevant role for TP53 in progression to the invasive phenotype [65].
Epigenetic mechanisms have been found to have a central role in both the earliest changes (i.e., atrophic gastritis and intestinal metaplasia) and the advanced stages of cancer [70, 71]. Studies on H. pylori and Epstein-Barr virus infection have shown that the carcinogenic effect of both these pathogens is reinforced by inducing methylation changes in the gastric mucosa [72–74]. The methylation status in the tumor tissue resembles the patterns found in serum samples, so methylation status has the potential to become a noninvasive oncological marker exploitable in the early diagnosis of gastric cancer, and a novel target for cancer prevention [75].
As in other human cancers, aberrant miRNA expression is a hallmark of gastric cancer [64]. Ueda et al. recently performed the largest study to date on gastric cancers [76]. Using a sizable number of gastric cancer tissues paired with nontumor samples, the authors identified 22 up- and 13 down-regulated miRNAs. Using the pattern of the 19 most significantly dysregulated miRNAs, it was also possible to discriminate gastric cancers according to their histological type. In particular, cluster analyses showed that miR-105, miR-100, miR-125b, miR-199b, miR-99a, miR-143, miR-145, and miR-133a were upregulated in diffuse-type gastric cancer, while miR-373-3p, miR-498, miR-202-3, and miR-494 were upregulated in intestinal-type lesions. Of note, miRNAs can be used as noninvasive biomarkers of gastric cancer [77], being readily and reproducibly detectable in various body specimens including blood, gastric fluids, feces, saliva, and others.
Gastric Cancer Secondary Prevention
Non-self-limiting chronic inflammation, mainly due to H. pylori infection [13, 19, 78], triggers both genotypic and phenotypic changes in the gastric mucosa. This process leads to an absolute loss of resident glands and/or to the native glands being replaced by inappropriate (metaplastic) glandular units (i.e., atrophic gastritis). The metaplastic variant of atrophy includes two main phenotypes: pseudo-pyloric metaplasia and intestinal metaplasia (IM). The atrophic transformation of the gastric mucosa provides the cancerization field in which (intestinal type) gastric cancer usually develops [27, 28, 78].
Metaplastic/atrophic glands are biologically “unstable” and prone to further dedifferentiation. This situation results in “neo-epithelia” harboring most of the biological traits of neoplastic cells. These (already) neoplastic epithelia lack the capacity for invasion, however, remaining topographically confined within the basal membrane of the glandular structure (i.e., intraglandular neoplasia (IGN); synonyms: intraepithelial neoplasia (IEN), noninvasive neoplasia (NiN); and formerly called dysplasia) [79]. Further progression of the molecular derangements, coupled with a proliferative advantage, loss of cell-to-cell adhesion, and the development of a capacity for invasion ultimately result in (early) invasive adenocarcinoma [50].
This natural history provides the rationale behind multidisciplinary strategies for cancer primary and secondary prevention [8, 28]. Several operative inconsistencies significantly influence efforts to anticipate gastric cancer detection, however, including: (i) the reliability of clinical/serological data used to assess gastric precancerous conditions; (ii) the endoscopic assessment of preneoplastic lesions; (iii) the biopsy sampling protocols applied; (iv) discrepancies in the histological classifications; and (v) interobserver variability in histology reporting.
In H. pylori-associated gastritis, atrophic changes are seen earlier in the angular (transitional) mucosa, involving the distal stomach only later on (i.e., antrally restricted atrophic gastritis), and finally spreading towards the (cranial) oxyntic mucosa (a condition sometimes called multifocal atrophic gastritis (MAG) , or atrophic pan-gastritis) [80]. It takes years to progress from nonatrophic inflammatory disease to its atrophic counterpart, consistently with the rising prevalence of atrophic gastritis with age. The distal-to-cranial spreading of atrophic changes can also be confidently considered an indication of a stepwise progression of the atrophic disease. Consistently with this hypothesis on the disease’s natural history, Japanese researchers identify oxyntic atrophy as the most advanced stage of H. pylori-associated gastritis.
Several studies have consistently associated the severity/topography of gastric atrophy with the risk of gastric cancer [8, 27, 28, 78, 81]. Built on the seminal experience of the Sydney System, Histological Division [82, 83], the histological phenotyping of gastritis implies a topography-based assessment of the inflammatory/atrophic changes involved. As a consequence, biopsy specimens should be obtained from each of the two mucosal compartments (e.g., three biopsy samples from the antrum—including the incisura angularis—and two from the gastric body) [19].
The “descriptive” philosophy behind the Sydney System has recently been replaced by a new approach to gastritis histology reporting [19]. The aim of the new diagnostic format (gastritis staging) is to enable a more definitive and clinically more readily perceptible stratification of the gastritis-associated risk of gastric cancer. The new staging format has yet to be included in the guidelines addressing the Management of Gastric Precancerous Conditions/Lesions (MAPS). These guidelines recognize the prognostic reliability of the staging approach, but base their operative recommendations on the topographical “spread” of atrophy/metaplasia alone, without considering the (more significant) prognostic message in gastritis staging [62].
Two related staging systems have been proposed and are in current clinical use: the OLGA and the OLGIM [84–86]. Both, OLGA and OLGIM, distinguish between four stages of gastritis (stages 0 to IV), associated with a progressively increasing risk of gastric cancer. The first staging system was presented in 2005 by an international group of pathologists and gastroenterologists (OLGA is an acronym for “Operative Link on Gastritis Assessment”) [86]. According to the OLGA approach, the stage of gastritis stems from the combination of the atrophy scores for the distal stomach with those assessed in the biopsy samples obtained from the proximal gastric mucosa [84]. This stage indicates the individual’s likelihood of developing a malignant neoplasia, and the vast majority of cases of cancer are expected to develop in patients in stages III and IV [87]. The stage of the organic lesions interestingly correlates with “functional” parameters of the gastric mucosa, and in particular with serum pepsinogens [88]. This correlation between “organic” and “functional” gastric disease may be of paramount importance when serologically selecting atrophic patients in whom endoscopy/biopsy procedures can be used as part of any gastric cancer secondary prevention effort.
A simplified staging system (OLGIM) focuses on the score/topography of intestinal metaplasia within the antral and corpus mucosa [85]. Which of the two staging approaches is more efficient in clinical practice, is still a debated issue; but both are consistent with the clinical priority of stratifying gastritis patients by their cancer risk [85, 89, 90]. Both systems identify stage III/IV patients as carrying a higher cancer risk, and consistently only associate the specific recommendation for endoscopy/biopsy surveillance with this (restricted) population. The prognostic value of gastritis staging, already recognized by the Maastricht IV Consensus Conference [91], has been recently confirmed by the “Kyoto Global Consensus Meeting on H. pylori gastritis” (Kyoto 2014).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:
 Lymphadenectomy—D1, D2, and D3
Lymphadenectomy—D1, D2, and D3
 The Role of Staging Laparoscopy and Peritoneal Cytology in Gastric Cancer
Radiation Treatment for Gastric Cancer
The Role of Staging Laparoscopy and Peritoneal Cytology in Gastric Cancer
Radiation Treatment for Gastric Cancer
 Methods of Reconstruction—BI, BII, Roux-en-Y, Jejunal Interposition, Proximal Gastrectomy and Pouch Reconstruction
Methods of Reconstruction—BI, BII, Roux-en-Y, Jejunal Interposition, Proximal Gastrectomy and Pouch Reconstruction
 Endoscopy and Endoscopic Ultrasound Examination of the Stomach
Endoscopy and Endoscopic Ultrasound Examination of the Stomach
 Pathology of Gastric Cancer
Pathology of Gastric Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


