Chapter 3 Oogenesis
INTRODUCTION
Oogenesis is an area that has long been of interest in medicine, as well as biology, economics, sociology, and public policy. Almost four centuries ago, the English physician William Harvey (1578–1657) wrote ex ovo omnia—“all that is alive comes from the egg.” Oogenesis exemplifies many fundamental biologic processes, such as mitosis, meiosis, and intracellular and intercellular signaling. The process of oogenesis begins with migratory primordial germ cells (PGCs) and results in an ovulated egg containing genetic material, proteins, mRNA transcripts, and organelles that are essential to the early embryo.
The generation of healthy eggs with the correct genetic complement and the ability to develop into viable embryos requires that oocyte development and maturation be tightly regulated. In an effort to describe in detail our current understanding of the basic biologic mechanisms behind this process, this chapter discusses mammalian oogenesis as it is understood in the mouse model (Fig. 3-1) and highlights clinical conditions that are related to defects in oogenesis.
PRIMORDIAL GERM CELLS
Primordial germ cells are the founder cells of the gametes. In many species, including Drosophila, Danio rerio (zebrafish), and C. elegans, germ cells are derived from germ plasm, which is maternally derived cytoplasm that contains germ cell determinants. In contrast, mammalian PGC fate is thought to be induced by cell–cell interaction requiring the adhesion molecule E-cadherin and signaling from surrounding tissues.1,2
The migration of PGCs from the base of allantois to the genital ridges has been mapped out by alkaline phosphatase staining; more recently, their migration in vivo has been visualized by green fluorescent protein (GFP) expressed under the control of a truncated Oct-4 promoter (GFP:Oct-4) acting specifically in PGCs.3,4 Primordial germ cells are derived extragonadally from yolk sac endoderm and part of the allantois that arises from the posterior part of the primitive streak.
Primordial germ cells can be distinctly identified at 7.5 days post-coitum (dpc, a measure of embryonic age) during gastrulation by their high expression of alkaline phosphatase or GFP:Oct-4. At 7.5 dpc, PGCs are seen at the root of the allantois, from which they migrate along the endoderm via passive transfer mechanisms, to arrive at the epithelial layer of the hindgut at 8 dpc. Then, with the acquisition of motility via long pseudopodia, they traverse through the wall of the hindgut at 9 dpc.3
Specific Genes Expressed for Migration of Primordial Germ Cells
Several genes are known to be required for PGC cell fate determination, migration, and survival in the mouse embryo. Bone morphogenetic protein 4 (Bmp4) and Bmp8b are maximally expressed in the extraembryonic ectoderm adjacent to the proximal epiblast, where precursors of PGCs are localized at 6.5 dpc.5,6 These two proteins are thought to have essential roles in the differentiation of PGC precursors into PGCs because PGCs are absent in both Bmp4 and Bmp8b homozygous null mutant mouse models.6,7
Bone morphogenetic proteins are members of the transforming growth factor β (TGFβ) superfamily of growth factors, which also includes the TGFβ family and activins. These signaling molecules have diverse and often critical functions during embryonic development and in adult tissue homeostasis. They signal via formation of heterodimers or homodimers of the highly conserved carboxyl-terminal domain, which is characterized by six or seven cysteine residues that are critical to the structure, stability, and function of these proteins. TGFβ superfamily members signal by binding to serine/threonine kinase cell membrane receptors that leads to phosphorylation of proteins of the Smad family (named after the C. elegans gene sma and Drosophila gene Mad, reviewed in Shi and Massague8).
Smad complexes can bind DNA and mediate transcriptional activation or repression. Smad1 and Smad5 are known to act downstream of the Bmp signaling pathway and may have important functions in PGC development because loss of these proteins results in decreased size of the PGC population.9–11
Migration of PGCs through the hindgut and along the mesentery is mediated by the binding of c-Kit, a tyrosine kinase receptor expressed in PGCs, to its ligand, stem cell factor (Scf). Scf is expressed by somatic cells and its gradient along the migratory path is thought to direct the migration of PGCs. Mutations in the dominant white spotting (W) and Steel loci, which encode c-Kit and Scf, respectively, result in defects in gametogenesis. Although PGCs form, they do not proliferate, migrate to ectopic sites, and aggregate prematurely, so that the genital ridges are poorly populated by germ cells and the animals are sterile.12,13
Interestingly, these mutant mouse models also exhibit defects in melanogenesis and hematopoiesis because these genes are also required for the migration of melanocytes and mast cells.14 PGCs also express integrin subunits, which may potentially interact with laminin and fibronectin produced by cells in the genital ridges during the formation of the gonads. For example, integrin β1 is required for PGC colonization of the gonads, as the migration of PGCs is arrested at the gut endoderm in integrin β1 null mutants.15
OOGONIA AND THEIR MEIOTIC ENTRY TO BECOME OOCYTES
Colonization of the genital ridges by PGCs occurs concomitantly with the formation of the ovaries, at 12 to 13 dpc. Postmigratory PGCs, together with somatic epithelial cells that have derived from the coelomic epithelium and mesenchyme, form the ovarian cortical sex cords. Germ cell survival beyond 13 dpc requires the C2H2-type zinc-finger transcription factor Zpf148, which may activate tumor suppressor protein p53 in germ cells at this stage of development.16 Around the same time, PGCs differentiate into oogonia, which are sexually differentiated germ cells that would undergo the last rounds of mitotic divisions to establish the germ cell population at 11 to 13 dpc.17
Our understanding of mammalian oogonium function has been limited, perhaps due to their transient existence. Incomplete cytokinesis during synchronous, mitotic divisions of the oogonia results in the formation of clusters, or cysts, that are connected by intercellular bridges 0.5 to 1.0 μm in diameter.17 Oogonia become oocytes by entering meiosis I at 14 dpc; by 17 dpc, most oogonia have transitioned to oocytes. In the perinatal period, these germ cell cysts undergo breakdown, after which only a third of the oocytes survive and become enclosed in a single layer of granulosa cells to form primordial follicles.17 Thus, perinatal germ cell apoptosis appears to be a developmental process that is differentially regulated compared to the loss of oocytes during follicular atresia that occurs in the sexually mature animal. Pepling and Stradling suggested that cyst formation and subsequent breakdown may serve to ensure the incorporation of mitochondria from dying oocytes into those that survive.17
Oocyte Development
Prophase of Meiosis I
In zygotene, homologous chromosomes pair and begin to synapse. Their synapses are maintained by the synaptonemal complex, which is formed by many protein subunits, including synaptonemal complex protein 3 (Scp3) in the axial elements, Scp1 in the central element, and Scp2.18 Synapsis is completed in pachytene and continues to be maintained until diplotene, when homologous chromosomes are held together mainly at sites of chiasmata. The crossing-over and recombination of chromosomes occur over 4 days in the pachytene stage, prior to the formation of ovarian follicles.
MutL and MutS Families of Proteins
DNA mismatch recognition and repair by the MutL and MutS families of proteins are well conserved from yeast to humans.19 MutL and MutS heterodimeric protein complexes are thought to interact to activate DNA mismatch repair. Although the MutL and MutS vertebrate homologs (Mlh and Msh, respectively) are generally known for their roles in maintaining genomic stability against tumor formation, gene targeting in mice revealed that Mlh1, Mlh3, Msh4, and Msh5 have essential functions in meiosis in both males and females.20–24
Mlh1 and Mlh3 homozygous mutant females have similar infertility phenotypes, in which the newborn ovaries appear normal with a normal number of follicles at various stages of development. However, there is a significant decrease in the number of oocytes that can complete meiosis I, meiosis II, and undergo subsequent development to two-cell embryos.21,23 Both Mlh1 and Mlh3 colocalize to chromosomes in pachytene and are thought to have critical functions in meiotic recombination, as supported by the decreased number of chiasmata formed in the oocytes of these mutant animals.21,23,25
Although oocytes seem to enter and arrest at diplotene, chromosomes are unpaired and cannot stably attach to the bipolar spindle, which leads to abnormal meiotic I spindle formation, abnormal or incomplete meiosis I, and fertilization failure.25 Further, in both human and mouse pachytene oocytes, MLH1 and MLH3 serve as molecular markers of recombination nodules, which are necessary for the crossover structures that are present in the diplotene stage.26
Similarly, Msh4 and Msh5 have essential functions in pachytene of prophase I, although their phenotypes were more severe.20,22,24 Female null mutants of Msh4 and Msh5 have significant loss of oocytes by postnatal days 2 to 4, before meiotic arrest at the diplotene stage.20,22,24 Their oocytes enter leptotene to form synaptonemal complexes but fail to undergo complete pairing at zygotene and fail to enter diplotene. These oocytes undergo apoptosis, which results in atretic follicles, loss of follicular architecture, and premature ovarian failure. Both Msh4 and Msh5 localize to chromosomes, can form heterodimers in vitro, and are thought to function at the same time point during chromosomal synapsis.22 The relevance of these findings from the mouse model to human reproduction is further supported by the expression of MSH4 and MSH5 in the human testes and ovaries.27
We do not yet know the exact mechanisms by which these Mlh and Msh proteins act or how they bind to chromosomes. The oocyte apoptosis phenotype has been attributed to failure of chromosome pairing or formation of synapses. However, recent genetic analyses based on the Spo11/Msh or Spo11/Mlh double null mutants indicate that oocyte apoptosis is a consequence of failure to repair double-strand breaks.28 Spo11 initiates double-strand breaks, which are required for recombination. In the absence of Spo11, the phenotype of Mlh1 and Msh5 was less severe and resembled that of Spo11. Similarly, Dmc1, another protein involved in recombination, and ataxia-telengiectasia mutated (Atm), a DNA damage checkpoint protein, are both required for the repair of double-strand breaks; in their absence, persistent double-strand breaks lead to oocyte death via apoptosis.28
The functional mechanism of a protein is often elucidated by identifying proteins with which it associates and the nature of their interactions. The colocalization of Mlh1 proteins with Scp3 and cyclin-dependent kinase 2 (Cdk2) may suggest interaction or related functions, especially since Scp3 and Cdk2 have also been shown to be required in meiosis.29,30 Scp3 is a key protein subunit in the synaptonemal complex, which holds homologous chromosome pairs to facilitate synapsis. Female mice lacking Scp3 are subfertile, because many of their oocytes contain univalents (unpaired chromosomes), exhibit abnormal chromosomal segregation, and produce a decreased number of viable embryos. Interestingly, their fertility worsens with age, so that this model is thought to be potentially powerful in studying the age-related subfertility and aneuploidy that are major problems in human reproduction.30
Cdk Family of Proteins
Cyclin-dependent kinase (Cdk2) belongs to the family of Cdk proteins, which are considered master regulators of the cell cycle.31 However, Cdk2-null mutants are unexpectedly viable; thus, Cdk2 is not required for mitosis during development in vivo to be required for prophase I of meiosis in the oocytes instead. Oocyte loss and premature ovarian failure occur by the first few postnatal days. Although the precise molecular defects differ, a similar phenotype is seen in the null mutants of Dmc1, Msh4, Msh5, and Atm, presumably because these oocytes fail to enter or complete pachytene, and enter the common apoptotic pathway by default. In contrast, by postnatal day 5, all oocytes in wild type mice have progressed to the diplotene stage, where they remain arrested until peri-ovulation in the sexually mature adult, when they resume and complete meiosis.
OOCYTE DEVELOPMENT AND EARLY STAGES OF FOLLICULOGENESIS
The transition from pachytene to diplotene marks the beginning of folliculogenesis, which is closely associated with subsequent oocyte development. Whereas no pachytene oocytes are found within follicles, approximately 80% of diplotene oocytes are enclosed in primordial or more developmentally advanced follicles.32 Primordial follicles are formed by the encapsulation of a diplotene-arrested oocyte by a single flat layer of granulosa cells. Subsequently, the granulosa cells, still in a single layer, assume a cuboidal shape to form primary follicles.
Transcription Factors
Our understanding of primordial and primary follicular development has recently been enlightened by the discovery of the requirement of two transcription factors at these stages of development (Fig. 3-2). Figα, a β-loop-helix-loop transcription factor, is required for oocyte survival into the primordial follicle stage.33 Based on its known role in the transcription of oocyte-specific genes zona pellucida 1 (zp1), zp2, and zp3, its role in primordial follicle formation is presumed to be the transcription of other oocyte-specific transcription factors.33
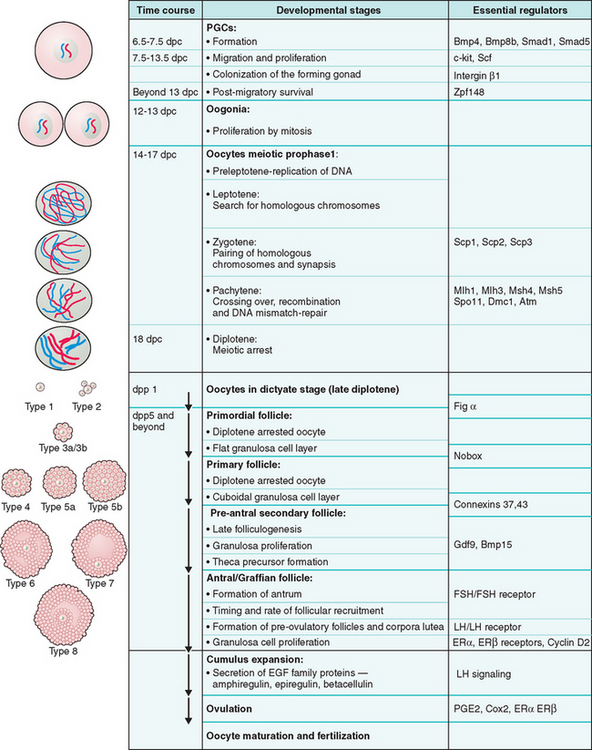
Figure 3-2 Schematic representation of oogenesis and folliculogenesis during development in the mouse model. This figure shows and correlates the developmental stages of the oocyte and follicle, and highlights examples of essential regulators at each stage. The “Types 1-8” nomenclature, as described by Pedersen and Peters191, is also shown to facilitate interpretation of the literature. PGCs, primordial germ cells; dpc, number of days postcoitum; dpp, number of days postpartum.
Another transcription factor, Nobox, which is an oocyte-specific homeobox protein, has a critical role in the transition of primordial to primary follicles.34 In the absence of Nobox, mouse ovarian follicles do not develop beyond the primordial stage, which results in massive oocyte loss in the early postnatal period.34 Identification of novel downstream target genes of Figα and Nobox in the future should provide insight into these early stages of folliculogenesis.
Zona Pellucida
The growing oocyte synthesizes zona pellucida glycoproteins, which comprise approximately 17% of the total cellular protein content. The zona pellucida proteins zp1, zp2, and zp3 are encoded by distinct genes and are coordinately expressed under the control of Figα during oocyte growth.33 These zona proteins form the zona pellucida or zona matrix that surrounds the growing oocyte, but each one has a critical role in proper oocyte and embryo development. zp1, the only zp protein that forms intermolecular disulfide bonds, contributes to 10% to 15% of the zona mass and is thought to provide structural integrity to the zona pellucida. zp3 serves as the receptor for sperm binding and the subsequent acrosome reaction; zp2 functions as a secondary receptor. Together, zp2 and zp3 mediate sperm binding and species specificity during fertilization.
These three glycoproteins are essential for fertilization and viability of growing oocytes and early embryos. Mice lacking zp3 cannot form the zona matrix and are sterile,35 and zp1 knockout mice have decreased fecundity and precocious hatching of early embryos from the structurally compromised zona matrix.36 Further, ectopic granulosa cells are seen between the oolemma and zona pellucida, and may increase the perivitelline space prior to ovulation.36 It is important to note that these may be four zona pellucida genes that are expressed in humans (ZP1, ZP2, ZP3, and zPB). Interestingly, abnormalities and lack of zona pellucida in human oocytes have been observed during IVF; however, the cause and implication of this defect in human reproduction is not well understood.
Regulation of Folliculogenesis by Oocyte Morphogens
Although bidirectional communication between the oocyte and follicular cells is required for the development of the meiotically competent oocyte, there is increasing support to view the oocyte as autonomous in determining its own fate by regulating follicular development via the secretion of various growth factors.37,38 The establishment of a morphogen gradient by the oocyte regulates follicular development via differential gene expression and function in granulosa cells. In the absence of these morphogens, follicle-stimulating hormone (FSH) induces all cumulus cells, which are directly adjacent to the oocyte and have distinct functions, to differentiate into membrane granulosa cells. Currently, several members of the TGFβ superfamily are the most well-understood candidate oocyte morphogens.
Transforming Growth Factor β
The mammalian oocyte expresses at least three TGFβ superfamily members: growth differentiation factor 9 (Gdf9), Bmp15, and Bmp6.38 Although Gdf9 is expressed throughout the reproductive tract and bone marrow,39 its oocyte expression is restricted to oocytes in follicles at the primary (type 3a), preantral, or more advanced stages of development.39,40 After fertilization, the level of Gdf9 transcripts decreases to low or undetectable in preimplantation embryos.41
Gdf9 is a critical regulator of follicular development. In Gdf9-deficient female mice, primordial and primary follicles appear normal, but development beyond type 3a follicles is blocked and the animals are sterile.42 Further, in the absence of Gdf9, theca cell precursors are absent around the follicles, and the expression of kit ligand and inhibin-α is upregulated in the granulosa cells of the primary follicles.43 The increased levels of kit ligand are thought to mediate excessive oocyte growth via interaction with the c-kit tyrosine kinase receptor on the oocyte, leading to abnormally large oocytes and their cell death.43 Interestingly, Gdf9 deficiency also causes failure of granulosa cells to proliferate or undergo cell death, which presumably leads to their abnormal differentiation into clusters of steroidogenic cells reminiscent of corpora lutea.43 Therefore, the oocyte can be viewed as the master regulator of early folliculogenesis via its secretion of Gdf9.
The oocyte expression pattern of Gdf9 is shared by its family member Bmp15, which is encoded by an X-linked gene.44,45 Bmp15 homozygous null mutant females are subfertile due to decreased rates of ovulation and early embryo survival. Further, an effect of gene dosage is observed in Gdf9+/− Bmp15−/− mice, which have a more severe phenotype than Bmp15−/− mice.46 In addition, cumulus–oocyte reconstitution and RNA interference (RNAi) experiments demonstrated that both proteins regulate cumulus expansion in vitro. Therefore, Gdf9 and Bmp15 are thought to have synergistic roles in the regulation of folliculogenesis.
Interestingly, naturally occurring mutations in GDF9 and BMP15 are common in certain strains of domestic sheep, perhaps as a result of breeding practices. Although sheep carrying mutations in both alleles of GDF9 or BMP15 are sterile, those carrying a single mutant allele have increased ovulation rates and are “super fertile.” Superovulation in the presence of heterozygosity of either of these genes is thought to be caused by decreased inhibin production by granulosa cells, which in turn leads to increased pituitary FSH secretion, development of more than one dominant follicle, and ultimately superovulation. Thus, the paradoxical increase in fertility when one allele is mutated exemplifies the impact of gene dosage on ovarian follicular function.45
RECRUITMENT OF OVARIAN FOLLICLES
Nongrowing mouse oocytes within primordial follicles have a diameter of approximately 12 μm and comprise the resting pool. Cohorts from the resting pool are continuously induced by ovarian paracrine signals to undergo coordinated oocyte and follicular growth during a process called initial recruitment (Fig. 3-347). The recruited, growing follicles are called primary follicles, but this growth phase is very protracted so that primordial and primary follicles may not be easily distinguishable. These growing follicles subsequently develop into secondary and antral follicles. (The timeline of different stages of follicular development in humans and rodents are shown in Fig. 3-4 and 3-5.47)

(From McGee EA, Hsueh AJW: Initial and cyclic recruitment of ovarian follicles. Endocrine Reviews 21:200–214, 2000; with permission. Copyright 2000, The Endocrine Society.47)

(From McGee EA, Hsueh AJW: Initial and cyclic recruitment of ovarian follicles. Endocrine Reviews 21:200–214, 2000; with permission. Copyright 2000, The Endocrine Society.47)

(From McGee EA, Hsueh AJW: Initial and cyclic recruitment of ovarian follicles. Endocrine Reviews 21:200–214, 2000; with permission. Copyright 2000, The Endocrine Society.47)
The oocyte grows from approximately 12 μm to approximately 80 μm in the fully grown state, which is achieved prior to formation of the antral follicle in sexually mature mice. In general, antral follicles tend to undergo follicular atresia, which is initiated by apoptosis in the granulosa cells, unless they are rescued by increased levels of FSH in the estrus or menstrual cycles in mice and humans, respectively. This cyclic recruitment of antral follicles results in the rescue and continued development of a few follicles, but usually only one dominant follicle prevails and develops to the periovulatory stage in the human menstrual cycle.47 Although cyclic recruitment is operative after puberty in humans, most antral follicles ultimately undergo atresia, which results in oocyte death.
Primordial follicles that have not been recruited are thought to remain dormant and be available for recruitment at a later time. Therefore, maintenance of the pool of nongrowing oocytes is necessary to sustain a normal reproductive life span. Müllerian inhibiting substance or antimüllerian hormone, a TGFβ superfamily member well-known for its regulatory role in the sexual differentiation of the reproductive tract during development, is expressed in granulosa cells of growing follicles starting in the perinatal period.48 During folliculogenesis, it inhibits the recruitment of primordial follicles to the pool of growing follicles by decreasing the responsiveness of granulosa cells to FSH.49 Antimüllerian hormone is recognized as an important clinical marker of the ovarian reserve of viable follicles.
Regulation of Folliculogenesis by Gonadotropins
Preantral follicle development, which involves oocyte growth with limited granulosa cell proliferation, is regulated predominantly by paracrine and autocrine signals. Although gonadotropins are not required for early follicle development in vivo, these follicles can develop in response to FSH in vitro.47 In contrast, gonadotropins are required for antrum formation and the rapid granulosa cell proliferation that results in the graafian or antral follicle.
FSHR comprises a large extracellular domain that binds the FSH hormone with high affinity, and a seven membrane-spanning domain that activates downstream cyclic adenylate cyclase signaling via the heterotrimeric G proteins.50 Mice deficient in the FSH β subunit, which normally forms heterodimers with the α subunits, are infertile due to arrested follicular development prior to formation of the antrum.51
Consistent with the critical role of FSH signaling via its receptor, follitropin receptor knockout (FORKO) mice are also infertile.52 At postnatal day 2, there are fewer nongrowing but more growing follicles, but by postnatal day 24, the numbers of resting and growing follicles are both decreased. Therefore, postnatal recruitment of resting follicles into the growing phase appears to be aberrantly accelerated, followed by arrest in further follicular recruitment later on.
Further, no antral follicles are observed. Although these mouse models confirm that FSH signaling is required for follicular development beyond the preantral stage, the phenotype of the FORKO mice also suggests that FSHR signaling is essential in the regulation of the timing and rate of follicular recruitment.52 In contrast, luteinizing hormone receptor knockout mice (LuRKO) females are infertile, presumably due to defects in the later stages of folliculogenesis because follicles up to the early antral stage are present, but preovulatory follicles and corpora lutea fail to form.53
FSH and FSHR-mediated downstream intracellular signaling results in gene and protein expression that exemplifies granulosa cell function. For example, antimüllerian hormone, which is implicated in primordial follicle recruitment, exhibits an aberrant expression pattern in FORKO mice.52 Although recruitment of theca cells and their expression of the LH receptor and P450 aromatase genes are independent of FSH signaling, expression of these genes in granulosa cells is dependent on FSH signaling.54 Similarly, the genes encoding for inhibin and activin subunits, whose homodimers and heterodimers are thought to be important paracrine and autocrine mediators of follicular growth, have decreased expression levels in granulosa cells in the FSHβ knockout model.54 Other genes that are dysregulated as a result of FSHβ deficiency include those encoding for androgen receptor (AR), estrogen receptor β (ERβ), and cyclin D2, all of which have been shown to be essential in the final stages of follicular development.54
Estrogen Receptors
Two estrogen receptors, ERα and ERβ, are expressed in granulosa cells in the antral follicle. Different knockout models have been generated for these two receptors based on different strategies of gene targeting, but they produced similar phenotypes.55 ERαKO females are infertile; ERβKO females are subfertile. In both ERαKO and ERβKO mice, folliculogenesis progresses normally until the large antral stage, when ERβ is critical for ER-mediated granulosa cell proliferation and subsequent ovulation.
In contrast, ERα is not critical for follicular growth but is required for ovulation. The ERαβKO females clarify the question of functional redundancy between the two ERs by demonstrating that in the absence of ERs, antral follicles only have single layers of granulosa and theca cells, multiple small pockets of antral fluid, and oocytes that are dissociated from cumulus cells.55
D-type Cyclin
Granulosa cell proliferation in late folliculogenesis also requires cyclin D2, which belongs to the D-type cyclin family of cell cycle regulators that promote the transition from G1 to S phases via binding to Cdk4 or Cdk6.56 Cyclin D2–deficient females have decreased FSH-mediated granulosa cell proliferation, which results in antral follicles that have significantly fewer layers of granulosa cells. This defect in granulosa cell proliferation also results in ovulation failure on stimulation by LH. However, the number of follicles is not decreased, luteinization of theca cells proceeds, and follicles differentiate to become corpora lutea.
We learn from the cyclin D2–deficient mutants that although these final stages of folliculogenesis and ovulation require cyclin D2 via FSHR-mediated activation, specifically of protein kinase A (PKA), oogenesis per se does not require these last rounds of granulosa cell proliferation. In fact, the oocyte not only appears normal, but it is also competent to undergo meiotic resumption and fertilization to produce viable embryos.56 Therefore, although oogenesis and folliculogenesis are closely intertwined, oocyte development is a cell-autonomous process to a certain extent, especially in the advanced stages and at periovulation.
THE ROLE OF CUMULUS CELLS IN OOGENESIS
Cumulus cells, also called corona radiata, are specialized granulosa cells that directly line the oocyte. In addition to their supportive role in cytoplasmic maturation of the oocyte, they have important functions in oocyte development, including the maintainenance of meiotic arrest and induction of ovulation. Ovulation is a broad term that encompasses the processes of follicular luteinization, follicle rupture, and meiotic resumption in the oocyte.
A critical step during LH-induced ovulation is the cumulus cell-mediated cumulus expansion. Mural granulosa cells express LH receptor, which allows them to respond to LH by secreting proteins of the epidermal growth factor (EGF) family—amphiregulin, epiregulin, and betacellulin—which are thought to act as paracrine signals that lead to cumulus expansion.57 During this process, cumulus cells disperse in the extracellular matrix, which comprises hyaluronic acid, tumor necrosis factor-stimulated gene 6 (TSG6), and serum-derived inter-α-inhibitor, all of which are essential for follicular rupture.58
The cumulus expression of Tsg6 and several other proteins implicated in cumulus expansion is regulated by prostaglandin E2 (PGE2) via its receptor EP2.58 Consistent with the critical role of PGE2 in cumulus expansion and ovulation, mice deficient in either EP2 or cycloxygenase-2 (Cox-2), which is the rate-limiting enzyme for PGE2, are infertile due to their inability to ovulate.58–60
Connexin and Gap Junctions
Cumulus cells mediate many of their important functions by communicating through gap junctions among themselves as well as with the oolemma. These gap junctions are intercellular channels formed by proteins of the connexin family for the diffusion of sugars, amino acids, lipid precursors, nucleotides, metabolites, and signaling molecules. Connexin family members share the same protein domains, including four membrane-spanning domains, two extracellular loops, a cytoplasmic loop, and cytoplasmic N- and C-terminals.61 In mice, there are at least 17 connexin proteins, whose distinct sequence or length in the cytoplasmic loops and C-terminal tails, as well as heterodimeric and homodimeric coupling, allow functional diversity.
In mice, connexin (Cx) 32, 37, 43, 45, and 57 are expressed in the cumulus–oocyte complex, where they are located between cumulus cells, on cumulus transzonal projections that anchor cumulus cells into the zona pellucida, or on the microvilli or plasma membrane of the oocyte.61
The critical role of gap junctions in oogenesis is exemplified by the sterility phenotype found in Cx37-deficient mice. Cx37 is made by both the oocyte and granulosa cells, but it may be the only connexin member in cumulus–oocyte gap junctions that is contributed by the oocyte.61 In the absence of Cx37, follicular development fails at the preantral-to-antral transition, so that most follicles are arrested at the primary follicle stage, and only a few small antral follicles are present. Further, ovulation does not occur despite the formation of numerous corpora lutea.62
Similarly, in vitro experiments show that Cx43, which is also expressed in granulosa cells, is required for folliculogenesis beyond the primary follicle stage.63 Most importantly, oocytes from these two mutant mouse models are meiotically incompetent, which may reflect the requirement of cumulus cell communication in the acquisition of meiotic competence.
These connexin-deficient experimental models demonstrate the critical role of gap junctions in folliculogenesis, but they do not explain the role of cumulus cells in the regulation of meiotic arrest and resumption in the oocyte. Numerous models and hypotheses attempt to explain how and to what extent cumulus cells mediate the high oocyte intracellular cAMP levels that are required to maintain meiotic arrest even after the oocyte has acquired meiotic competence.64 Although the complete story remains to be told, several key players have been identified.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


