Fig. 9.1
Liver development. (a) 30-day embryo. The caudal portion of the hepatic diverticulum (HD) forms the extrahepatic biliary tree (green), while the cranial portion penetrates the septum transversum and the endodermal cells differentiate into hepatoblasts (cells in yellow). (b) Development and remodeling of the ductal plate. Pluripotential hepatoblasts differentiate into hepatocytes (brown) and ductal epithelium (green) at the limiting plate (1). The biliary-type cells form a continuous layer at the periphery of the portal tracts (the ductal plate) (2), which later becomes a double-layer structure (3) and subsequently develops a central lumen (4). Definitive bile ducts form and become more centrally located as excessive epithelium undergoes remodeling by apoptosis and growth of mesenchyme (5, 6). GB gall bladder, HD hepatic duct
The pre-hepatocytes or hepatoblasts proliferate within the septum transversum, organized in cords around developing sinusoids derived from branches of the vitelline veins that penetrate the septum transversum (MacSween et al. 2002; O’Rahilly and Muller 2001). The early hepatocyte cords that form around the developing sinusoids remain thick throughout intrauterine life and for the first few months after birth, thinning to two-cell trabeculae by 5 months of life and adopting the single-cell cord architecture of the adult during the first 5 years. Development of the hepatic primordium is dependent on continuous interactions with the adjacent mesoderm and on the concerted development of the hepatic vasculature, as revealed by studies of Klk1 -/- mouse embryos, which lack mature endothelial cells, with consequent inhibition of expansion of the liver bud (Zhao and Duncan 2005). Growth of the liver bud is very rapid between the 4th and 10th weeks of gestation in humans, such that by 50 days, 700 portal branches from the second to the sixth order can be counted (Collardeau-Frachon and Scoazec 2008). In summary, the mechanisms controlling liver growth appear to be well conserved among various organisms and depend on the coordinated expression of transcription and signaling factors from the cardiac mesoderm, the septum transversum, and the endoderm to commit the endodermal cells to a hepatic fate and to induce and regulate hepatic gene expression. This growth requires the action of a large number of factors and downstream cascades, such as hepatocyte growth factor (HGF), transforming growth factor-β (TGF-β), tumor necrosis factor-alpha (TNF-α) and the Wnt/β-catenin pathway, which involve both proliferation and apoptosis of the hepatoblasts. A full account of this rapidly evolving subject is beyond the scope of this chapter and is the subject of several excellent reviews (Lemaigre 2009; McLin and Zorn 2006).
The vascular architecture of the liver is unique in that the organ receives both a venous blood supply via the portal vein from the intestines, pancreas, and spleen, responsible for its functional circulation, and an arterial supply conveying nutrition and oxygenated blood from the hepatic artery. The development of the liver is closely associated with changes in the omphalomesenteric (vitelline) veins and in the umbilical veins. The definitive venous pattern within the liver is established by the 7th week (Saxena et al. 2003). Selective involution of this venous network during the first few weeks of development results in obliteration of segments of the vitelline veins and joining of the umbilical veins to the vitelline venous system (Corness et al. 2006). The portal vein arises from anastomotic channels derived from the paired vitelline veins in the hepatic primordium. The intrahepatic segments of the portal vein and its branches become surrounded by mesenchyme, from which portal spaces will develop and within which arterial segments will grow. The proximal end of the right vitelline vein forms the terminal part of the inferior vena cava (IVC), its remnants in the liver become the hepatic venous system, and its distal portion will form the splenic and superior mesenteric veins. The left vitelline vein forms part of the inferior vena cava. By the 6th week, the right umbilical vein disappears, and all oxygenated blood from the placenta enters the liver from the left umbilical vein. After the umbilical vein enters the liver, it gives off numerous branches to the left lobe before it drains into the ductus venosus, an oblique channel formed by the coalescence of some of the hepatic sinusoids that connect the umbilical vein to the inferior vena cava, diverting a portion of the oxygenated blood directly to the heart (Fig. 9.2).
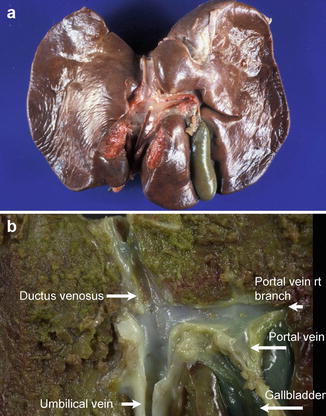
Fig. 9.2
The ductus venosus in the fetal liver. (a) A catheter in the umbilical vein points toward the ductus venosus, which allows blood from the umbilical vein to drain directly into the inferior vena cava during fetal life. (b) Cut section of the liver from a 29-week-old fetus further illustrates the relationship between the ductus venosus, umbilical vein, and portal vein. The left lobe of the liver is to the left of the picture
Hemodynamic studies performed during intrauterine life have confirmed that, until birth, the umbilical vein is the predominant liver vessel, and that the left lobe is supplied mainly by oxygenated blood from the umbilical vein, while the right lobe of the liver receives blood predominantly from the portal vein (Collardeau-Frachon and Scoazec 2008). This vascular asymmetry is believed to account for differences observed between the right and left lobes of the liver in stillbirths and neonates (Ghosh and Emery 1973; Emery 1955). At birth the left lobe is comparably larger than in later life. Birth results in the closure of the umbilical circulation, starting with collapse of the umbilical arteries, followed by closure of the umbilical venous flow, the latter complete by 15–20 days. This decrease in blood flow results in collapse of the ductus venosus and its obliteration by fibrous tissue. The umbilical vein becomes the ligamentum teres, whereas the ductus venosus transforms into the ligamentum venosum. Fetal blood flow through the hepatic artery is insignificant compared with that delivered by the umbilical and portal veins. The development of the hepatic arterial system parallels that of the bile ducts and reaches maturity at approximately 15 years of age (Saxena et al. 2003).
The liver becomes a site of hematopoiesis between weeks 6 and 7. Intense hematopoietic activity is present diffusely in the midtrimester through 24 weeks of gestation. As the fetus enters the third trimester, hematopoiesis diminishes and becomes focal. Little hematopoiesis is seen in portal tracts after 32 weeks of gestation, and by 36 weeks, hematopoiesis remains only as scattered islands in the hepatic parenchyma (Fig. 9.3).
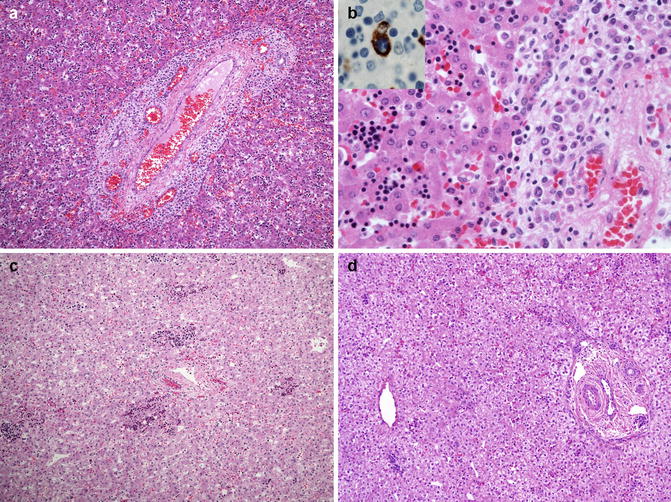
Fig. 9.3
Hematopoiesis in the fetal liver. (a) Hematopoietic activity is marked in the liver of an 18-week fetus. (b) Higher-power view of A demonstrates erythropoiesis in the sinusoids and myelopoiesis in the portal tract to the right of the illustration. Inset. CD68-staining macrophage engulfing and surrounded by erythroid cells in a hepatic sinusoid, representing a so-called Bessis island. (c) By 28 weeks, erythropoiesis has diminished and myelopoiesis is largely inapparent. (d) In this 36-week fetus, erythropoiesis is present only as scattered small islands in the sinusoids
9.1.2 Extrahepatic Biliary Tree
As mentioned above, the extrahepatic biliary tree is derived from the caudal part of the hepatic diverticulum as the latter elongates and widens. This portion of the endoderm is closely associated with the ventral pancreatic bud, and gene expression maps have identified a number of transcription factors that are common to the epithelial cells lining the developing ducts and to the pancreas and duodenum (Pdx-1, Prox-1, HNF-6). Mice deficient in these factors exhibit abnormalities of the biliary tree and gallbladder with abnormal differentiation toward a pancreatic phenotype (Lemaigre 2009; Strazzabosco and Fabris 2012). The morphogenesis of the biliary tree depends on close interaction with the surrounding mesenchyme. By the 4th week post-fertilization, the cystic duct and gallbladder are visible as a bud on the hepatic diverticulum (Fig. 9.1a). By the 5th week, the gallbladder, cystic duct, hepatic ducts, common bile duct, and pancreatic duct are well demarcated. The gallbladder elongates but remains tubular until the 11th week when it dilates into a saccular shape (Tan and Vijayan 2001). The distal common bile duct enters the duodenum where it is joined by the duct from the ventral pancreas. This junction initially lies outside the duodenum until the 8th week of gestation at which time it migrates into the duodenal wall. Failure of this process may result in an abnormally long choledocho-pancreatic channel associated with choledochal cyst formation. The major bile ducts at the porta hepatis are fully formed by the 11th week of gestation. Initially believed to be a solid cord of cells, more recent studies, such as those by Tan (Adeva et al. 2006), suggest that the anlage of the extrahepatic biliary tree has a continuous patent lumen, which is visible, according to these authors, as early as 29 days post-fertilization. The most cranial portions of the hepatic ducts would appear to be in direct continuity with converging ductules at the hepatic hilum, which in turn are continuous with the ductal plate and the developing intrahepatic ducts (Nakanuma et al. 1997). These observations would preclude the possibility that biliary atresia results either from a failure of “canalization” or from nonunion of separately developing cranial and caudal portions of the biliary tree. Although much less is known about the molecular mechanisms responsible for the development of the extrahepatic than the intrahepatic biliary tree, murine models deficient in Pdx-1 or Hes-1 (a Notch-dependent transcription factor), HNF6, HNF-1b, or Foxf1 (a transcription factor target for the sonic Hedgehog signaling) have an altered development of the gallbladder and of the common bile duct (Strazzabosco and Fabris 2012).
9.1.3 Intrahepatic Biliary Tree
The development of the intrahepatic bile ducts (IHBDs) begins between the 5th and 9th week post-fertilization. The biliary epithelium is believed to arise from precursor bipotential hepatoblasts which can differentiate into either hepatocytes or biliary epithelial cells, also referred to as cholangiocytes (Spagnoli et al. 1998). Beginning around the 6th week, the hepatoblasts adjacent to the developing portal tract mesenchyme flatten and become a continuous layer of biliary-type cuboidal cells, the ductal plate (Fig. 9.1b). At the hilum, the ductal plate is in direct continuity with the developing hepatic duct, and its remodeling leads to the formation of the intrahepatic portion of the right and left branches of the hepatic duct (Tan, 1994 #2649). The differentiation of the hepatoblasts into cells destined for the biliary tree and cells destined for the hepatic parenchyma appears to be determined at the time of ductal plate formation. Immunohistochemical studies have illustrated the development of the intrahepatic bile ducts, exploiting the differences in cytokeratin (CK) subtype expression between hepatocytes and biliary epithelial cells: precursor hepatoblasts express CK 8, 18, and 19; mature hepatocytes express CK 8 and 18 but not 19, whereas biliary epithelium expresses CK 8,18, 19, and 20. CK7, in the authors’ experience, is not as robustly expressed as CK19 in biliary epithelium until the third trimester. Around weeks 6–7 a prominent rim of CK19+ cells forms along the outer boundary of the developing portal tracts, forming the so-called ductal plate (Desmet 1998; Van Eyken et al. 1988). During the next few weeks, the rim becomes a continuous double-layered saccular sleeve around each portal tract (Fig. 9.4). Remodeling of the ductal plate, starting around week 12, is a dynamic process weaving together loss of continuity of the epithelial sleeve with the formation of discrete tubular spaces, the incorporation of the primitive tubules more centrally into the portal tract mesenchyme, and the elimination of excess epithelial elements. This tubulogenesis is transiently asymmetrical, driven by cholangiocyte-like cells on the portal tract side, and by hepatocyte-like cells on the parenchymal side of the ductal plate (Strazzabosco and Fabris 2012). The ductal structures gradually assume a symmetrical arrangement as the hepatoblasts are replaced by cholangiocytes. This process is believed to proceed from the hepatic hilum and extend centrifugally toward the periphery of the liver, resulting in a continuous development of IHBDs throughout fetal life, and appears to continue into the first month of life ex utero (Crawford 2002). Thus, in later stages, histologic examination of the fetal liver reveals more mature ducts in the hilar region, while the ducts in the periphery are still in ductal plate configuration (Desmet 1998) (Fig. 9.5). The development of the intrahepatic biliary tree is under the control of numerous transcription factors, including HNF6 and HNF1β (Strazzabosco and Fabris 2012). In addition, Notch-2 and JAG1 appear to also play key roles in biliary development, exemplified by paucity of bile ducts seen in Alagille syndrome resulting from mutations in JAG1. Studies in mice have shown that Jagged-1 is expressed by portal mesenchymal cells and interacts with Notch-2 expressed in hepatoblasts at the ductal plate. Inactivation of Jagged-1 in mesenchymal cells results in defective maturation of bile ducts in mice (Hofmann et al. 2010). Studies using resin casts in mouse models have shown that the three-dimensional density of the intrahepatic biliary tree appears to be regulated by Notch gene dosage during development (Sparks et al. 2010).
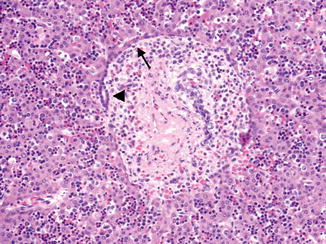
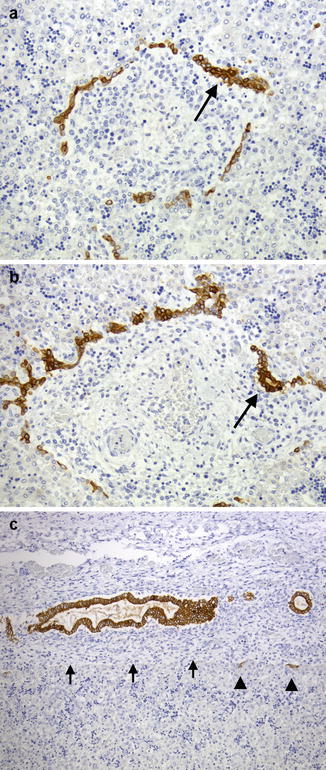
Fig. 9.4
Ductal plate development in a 16-week fetus. The ductal plate first develops a double layer of cells (arrow), and then a central lumen forms between these two layers (arrowhead)
Fig. 9.5
Ductal plate development in a 24-week fetus. CK19 immunostain. Comparison of peripheral areas (a, b) and areas near the hilum (c) in the same liver demonstrates the spectrum of ductal plate remodeling. Early changes of ductal plate development characterized by focal duplication (arrow, a) and early duct formation (arrow, b) are seen in the periphery, while near the hilum, ducts are more centrally located as the ductal plate elements disappear (arrowheads, c) at the limiting plate (arrows)
The intrahepatic biliary system is not mature at the time of birth, with expression of canalicular transporters present by mid-gestation, but with a different immmunohistochemical pattern than in adults (Chen et al. 2005). Development of the intrahepatic bile ducts also continues after birth, and remodeling of the ductal plate in the most peripheral portal tracts is probably not complete until 6 weeks after birth (Van Eyken et al. 1988); this has implications for the diagnosis of bile duct paucity on a liver biopsy in a premature infant. The adult liver, weighing approximately 1,400 g, is more than 10 times the weight of the newborn liver, but information on the development and growth of the liver after birth is scanty. Landing and Wells (1991) proposed that the postnatal human liver grows by an increase in the number of peripheral lobules with associated branching and elongation of the portal tracts. The intrahepatic tree can be divided into large bile ducts (300–800 μm in humans), which include hepatic, segmental, and area ducts, and small bile ducts (<300 μm), which include septal bile ducts (100–300 μm), interlobular bile ducts (15–100 μm), and bile ductules (diameter <15 μm) (Strazzabosco and Fabris 2008). The number of orders of intrahepatic branches in the fetus or newborn is not yet known (Crawford 2002), while according to Ludwig (Beuers and Sackmann 1998), 10–12 orders of branches are present in the adult liver. The large ducts have associated mucous peribiliary glands and can be grossly identified, while the small ducts are not associated with peribiliary glands and are identifiable only by microscopy. The interlobular (or terminal) bile ducts branch into bile ductules, which seem to have both an intraportal and periportal segment and are entirely lined by cholangiocytes and invested by a thin layer of mesenchyme. These ductules collect bile from the canals of Hering, which are lined in part by hepatocytes and in part by cholangiocytes and represent the link between the hepatocyte canaliculi and the biliary tree. Canals of Hering appear as small clusters of cells positively stained by cholangiocyte immunomarkers (cytokeratin 7 or 19), located at the periphery of the portal tracts and periportal region (Roskams et al. 2004) (Fig. 9.6). Canals of Hering and bile ductules appear to house the progenitor cells that will proliferate and result in the ductular reaction of chronic liver disease. Cholangiocytes possess primary cilia, which play an important role in regulating their biological activities. Cholangiocyte cilia also contain proteins such as polycystin and fibrocystin, defects of which are involved in biliary dysgenesis (Strazzabosco and Fabris 2008). Cholangiocytes exhibit functional heterogeneity depending on their location in the biliary tree: those in the larger bile ducts play a role in bile production via both absorptive and secretory functions, whereas cholangiocytes in the smaller branches, such as bile ductules and canals of Hering, have the ability to participate in inflammatory processes and to act as progenitor cells (Strazzabosco and Fabris 2008).
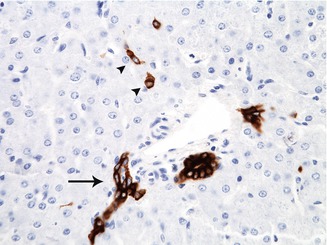
Fig. 9.6
A bile ductule at the edge of the portal tract (arrow) stained by immunohistochemistry with cytokeratin 19 appears as a small cluster of cells which extends into the surrounding lobule. Canals of Hering, which link the canaliculi with the biliary tree, appear in this microphotograph as isolated cells in the periportal area (arrowheads)
The peribiliary glands are histologically divided into intramural and extramural structures. The former consist of simple tubular glands with much mucin and are sparsely and irregularly distributed within the ductal wall. The latter are characterized by the presence of excretory units that consist of seromucinous acini and a conducting system in the periductal tissue. Pancreatic exocrine acini are occasionally admixed with extramural glands. These peribiliary glands appear in the late fetal period and complete their development about 15 years after birth along with the peribiliary capillary plexus. Extramural and intramural glands secrete neutral and acid mucin into the ductal lumen. Extramural glands contain several enzymes for digestion of protein and lipids. Neural and vascular supply of these glands may be related to the regulation of their secretion. Specific and nonspecific immune responses within this glandular system may also be essential in the sterility of bile (Nakanuma et al. 1994).
9.2 Developmental Anomalies
9.2.1 Liver
9.2.1.1 Agenesis of the Liver
Agenesis of the liver is incompatible with life and has been reported in stillborn fetuses with other severe anomalies (Ishak and Sharp 2002). Hypoplasia or agenesis of one lobe is rare and usually involves the right lobe. It is usually accompanied by a retrohepatically or suprahepatically located gallbladder. Many cases are discovered incidentally. Others develop biliary tract disease or portal hypertension (Kanematsu et al. 1991; Makanjuola et al. 1996). Significant hypoplasia or absence of the right lobe associated with malposition of the gallbladder may cause compression of the cystic duct resulting in choledocholithiasis and cholecystolithiasis. Hypoplasia of the left lobe is rare and usually found incidentally at autopsy.
9.2.1.2 Anomalies of Position
Anomalies of position are usually the result of laterality abnormalities (situs inversus and asplenia-polysplenia syndromes), diaphragmatic defects, or abdominal wall defects. Situs anomalies have been associated with the so-called embryonal form of biliary atresia, usually in combination with splenic and other laterality defects.
9.2.1.3 Accessory Liver Lobes
Accessory liver lobes can be present in the inferior surface of the liver (Ishak and Sharp 2002). Very rarely, an intrathoracic accessory lobe has been reported. These come to clinical attention because they can mimic a tumor (Kitamura et al. 2012) or may torse if on a pedicle, resulting in an abdominal emergency (Umehara et al. 2009). Riedel’s lobe is a downward projection of the right lobe of the liver and is an anatomic variation rather than a true accessory lobe (Gillard et al. 1998). It is usually asymptomatic but may be mistaken for a tumor or be the site of a metastasis or a hepatocellular carcinoma.
9.2.1.4 Ectopic Liver
Ectopic liver is defined as an anomalous location of the liver tissue without connection to the main mass (Collan et al. 1978). Ectopic liver has been described in the falciform ligament, lung, spleen, retroperitoneum, adrenal gland, and omentum; the most common site appears to be the gallbladder. Several case reports have also described ectopic liver within the umbilical cord (Vaideeswar et al. 2011).
9.2.1.5 Heterotopias
Heterotopias in the liver include adrenal, pancreatic, splenic, and thyroid heterotopias. Intrahepatic splenosis occurs after trauma or surgery; it can mimic a tumor and may result in misleading imaging during radiologic investigations (De Vuysere et al. 2000).
9.2.1.6 Congenital Absence of the Portal Vein
Congenital absence of the portal vein is a rare vascular malformation. In that condition, blood carried by the splenic and superior mesenteric drains into a systemic vein, resulting in an extrahepatic portosystemic shunt. Intrahepatic portosystemic shunts, by contrast, arise from an aberrant connection between the portal venous system and the hepatic veins. In a review of 16 cases, agenesis of the portal vein was associated with other congenital anomalies, including cardiac defects, skeletal anomalies, and polysplenia, and with benign or malignant hepatic neoplasms (Guariso et al. 1998).
9.2.1.7 Hypoplasia of the Portal Vein
Hypoplasia of the portal vein often occurs in patients with biliary atresia (Hwang et al. 2013). Along with atresia and stenosis, hypoplasia of the portal vein results in portal hypertension.
9.2.1.8 Cavernous Transformation of the Portal Vein
Cavernous transformation of the portal vein is a spongy trabeculated venous lake which replaces the portal vein, resulting from the development of collateral venous channels around a stenosed or occluded portal vein. It is a major cause of portal hypertension. Two theories explaining the pathogenesis have been proposed. Cavernous transformation may be secondary to portal vein thrombosis due to umbilical vein catheterization or infection, or it may represent an angiomatous malformation.
9.2.2 Biliary Tree Anomalies
9.2.2.1 Isolated Agenesis of the Gallbladder
Isolated agenesis of the gallbladder is a rare anomaly. Its incidence is reported to be 10–65 per 10,000 and is more common in females (ratio 3:1) (Kasi et al. 2011). Bennion et al. (1988) described three categories: agenesis in association with multiple fetal anomalies, asymptomatic (agenesis discovered either at autopsy or at laparotomy for an unrelated diagnosis), and symptomatic (typically during the fourth to fifth decade).
9.2.2.2 Duplication of the Gallbladder
Duplication of the gallbladder is somewhat more frequent, and its anatomy ranges from a septate gallbladder to bilateral gallbladders with separate cystic ducts (well reviewed in (Skandalakis et al. 1994)). These are usually discovered at the time of laparotomy for cholecystitis.
9.2.2.3 Accessory Hepatic Ducts
Accessory hepatic ducts emptying into the cystic duct (cystohepatic ducts) or gallbladder (cholecystohepatic ducts), with a connection to a part of the liver, have been observed with an incidence rate varying from 1 to 30 % in different series (Narasimhan et al. 2001). They provide biliary drainage in some cases of obstruction (Champetier et al. 1991; Jamshidi et al. 1999; Kihne et al. 1980; Pietsch et al. 1978). These may be missed during preoperative radiologic investigations and discovered only at surgery (Harish et al. 2001).
9.2.2.4 Duplication of the Common Bile Duct
Duplication of the common bile duct is extremely rare. The majority of cases have been reported in the Japanese literature (Yamashita et al. 2002). The age at diagnosis has ranged from 4 days to 80 years, with a mean of 46.1 years. Symptoms (pain) are secondary to inflammatory processes around the biliary system (cholangitis, cholecystitis, cholelithiasis, liver abscess, and pancreatitis) or cancer of the biliary system, pancreas, or stomach. Cancer is a frequent complication when duplication is associated with pancreatobiliary maljunction (union of the pancreatic and biliary ducts outside the duodenal wall). The accessory common bile duct may open into the stomach, duodenum, or pancreatic duct or be separated by a septum from the common bile duct. Gastric cancer has only been reported in cases of an accessory duct opening into the stomach.
9.2.3 Fibrocystic Diseases of the Liver/Ductal Plate Malformation
Fibrocystic diseases of the liver encompass a heterogeneous group of disorders whose characteristic hepatic lesions include fibrosis (in most instances), with biliary dysgenesis, most frequently manifested as the ductal plate malformation (Table 9.1). The ductal plate, as described in the previous section, consists of a sleeve-like layer of cells that surround the developing portal tracts and will give rise to the intrahepatic bile ducts. This process involves focal dilatation of the original ductal plate to form tubules followed by involution of the intervening non-tubular elements. Arrest or derangement in remodeling leads to persistence of the primitive bile duct configuration, for which the term ductal plate malformation (DPM) was coined by Jorgensen (1977). The DPM is characterized by an increased number of irregular biliary channels in a circinate arrangement at the periphery of the portal tracts (Fig. 9.7). These channels appear to branch or anastomose as they form irregular profiles with angular shapes that have a tendency to become dilated or even cystic. The ductal plate derivatives are always a continuous structure during development are in communication with the biliary tree, and thus can become cystic. The epithelial lining consists of a single layer of low columnar to cuboidal cells. Focally, the abnormal ducts have intralobular extensions where the basement membrane of the epithelium appears to be in direct contact with the hepatocytes.
< div class='tao-gold-member'>
Table 9.1
Disorders associated with congenital hepatic fibrosis/ductal plate malformation
Syndrome | Inheritance (OMIM number) gene, location |
|---|---|
Autosomal dominant polycystic kidney disease | AD (173900) |
Genetically heterogeneous | |
Polycystin 1, 16p13 | |
Polycystin 2, 4q21–23 | |
Autosomal recessive polycystic kidney disease | AR (263200) |
Fibrocystin, 6p21.1-p12 | |
Bardet-Biedl | AR (209900) |
Genetically heterogeneous | |
Oligogenic inheritance reported | |
BBS1, 11q13; BBS2, 16q21; BBS3, 3p13; | |
BBS4, 15q24.1 | |
BBS5, 2q31; BBS6, 20p12; BBS7, 4q27; | |
BBS10, 12q21.2 | |
BBS12, 4q27; MKKS, 20p12.2 | |
Caroli’s syndrome | AD (600643) |
Cumming campomelia | AR (211890) |
Elejalde | AR (256710) |
May be same entity as Griscelli syndrome caused by mutations in the MYO5A gene | |
Ellis-van Creveld (chondroectodermal dysplasia) | AR (225500) |
EVC, EVC2, 4p16 | |
Glutaric aciduria type II | AR (231680, 231675) |
Genetically heterogeneous | |
ETFA, type IIA 15q23–q25; ETFB, type IIB, 19q13.3 | |
ETFQO, type IIC, 4q32 | |
Jeune – asphyxiating thoracic dystrophy | AR (208500), 15q13 |
Joubert syndrome-related disorders (including COACH syndrome) | AR (216360) |
Heterogeneous | |
CC2D2A, 4p15.32; TMEM67, 8q22.1; RPGRIP1L, 16q12.2 | |
Juvenile nephronophthisis | AR (256100, 266900) |
Genetically heterogeneous | |
NHP1 (nephrocystin-1), 2q13; other loci | |
Mannose phosphate isomerase deficiency (congenital disorder of glycosylation type IB) | AR (602579) |
MPI, 15q24 | |
Meckel-Gruber | AR (249000) |
Genetically heterogeneous | |
MKS1, 17q22–23; MKS2, 11q; MKS3, 8q; and others | |
Polycystic liver disease | AR (174050) |
PRKCSH, 19p13.2; SEC63, 6p21 | |
Renal-hepatic-pancreatic dysplasia (Ivemark) | AR (208540) |
NPHP3, 3q22.1 | |
Senior-Loken | AR (266900) |
Genetically heterogeneous | |
NPHP1, 2q13; NPHP4, 1p36; NPHP5, 3q21 | |
NPHP6, 12q21; SDCCAG8, 1q44. | |
Short rib-polydactyly, type I (Saldino-Noonan) | AR (263530) |
Short rib-polydactyly, type II (Majewski) | AR (263,520) |
Syndrome with features of Joubert, Meckel, and Smith-Lemli-Opitz syndromes | AR (213010) |
Trisomy 9 | Chromosomal |
Tuberous sclerosis | AD (191100) |
Hamartin, TSC1, 9q34 | |
Tuberin, TSC2, 16p13.3 | |
Zellweger cerebro-hepato-renal | AR (2141000) |
Peroxisomal disorder, genetically heterogeneous | |
PEX1, 7q21–22; PEX2, 8q21.1; PEX3, 6q23–24 | |
PEX5, 12p13.3; PEX6, 6p21.1; PEX12, 17 | |
Other loci |
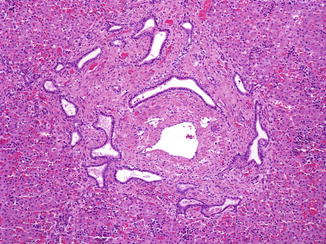
Fig. 9.7
Ductal plate malformation. A 1-day-old, 38-week gestation male infant with autosomal recessive polycystic kidney disease. Irregular ductal profiles with angular shapes and focal dilatation are seen mostly at the periphery of portal tracts
Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


