negative predictive values are 100% and 91%, respectively. Negative CT scans often detect other abnormalities including appendicitis, pelvic inflammatory disease, diverticulitis, abdominal aortic aneurysm, and bladder cancer. Plain abdominal radiography assesses whether the stone is radiopaque, which is the case 75% to 90% of the time. Ultrasonography has a high specificity but a much lower sensitivity than CT. Ultrasonography is appropriate as the initial imaging test in pregnancy and in pediatrics and in patients who should avoid radiation. Intravenous pyelography has been replaced by helical CT as the preferred imaging test.16 Secondary signs of urinary tract obstruction such as ureteral dilatation, hydronephrosis, and perinephric stranding, are variably seen depending on the duration of pain prior to imaging and the sign itself.17
TABLE 20.1 Urinary Chemistries in Evaluation of Nephrolithiasisa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
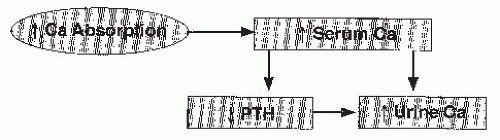 FIGURE 20.1 Pathogenesis of absorptive hypercalciuria. Intestinal hyperabsorption of calcium leads to an increase in serum calcium and a reduction of parathyroid hormone (PTH). The increase in the filtered load of calcium and the reduced calcium reabsorption produced by loss of PTH activity lead to hypercalciuria. |
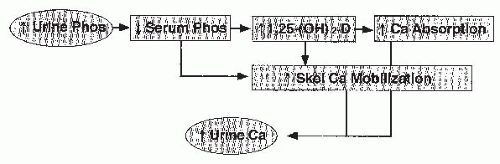 FIGURE 20.2 Renal phosphaturia. Hypophosphatemia leads to increased production of 1,25(OH)2D3, intestinal hyperabsorption of calcium, and increased skeletal calcium mobilization. As a result, hypercalciuria develops. |
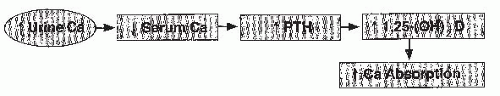 FIGURE 20.3 The pathogenesis of renal hypercalciuria. Defective renal calcium reabsorption leads to hypocalcemia and a stimulation of parathyroid hormone (PTH) secretion. The latter increases the production of 1,25(OH)2D3, stimulating calcium absorption and leading to hypercalciuria. The hypercalciuria in turn compensates for the reduction in serum calcium but compensation never completely restores normal calcium, and elevated PTH values are required in this syndrome. |
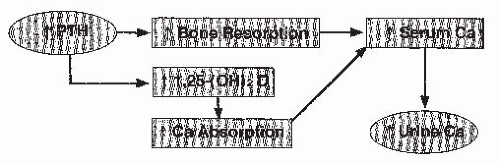 FIGURE 20.4 Pathogenesis of hypercalciuria and primary hyperparathyroidism. Excess secretion of parathyroid hormone (PTH) stimulates bone remodeling and elevations of 1,25(OH)2D3 and calcium absorption. The resultant increase in serum calcium leads to an increase in the filtered load of calcium.The latter overwhelms the stimulation of renal tubular calcium transport by PTH, and hypercalciuria results. |
suppressed and the rarity of secondary hyperparathyroidism call for a reexamination of the issue regarding renal tubular calcium fluxes.
TABLE 20.2 Intestinal Calcium Absorption in Normal Subjects and Patients with Idiopathic Hypercalciuria | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
with the CLCN-5 mutations involves impaired solute reabsorption by the proximal tubule and range from kaliuresis, glycosuria, phosphaturia, and/or hypouricemia. Constant findings of Dent disease include hematuria and low molecular weight proteinuria. Dent disease progresses to renal failure due to nephrocalcinosis and tubulointerstitial nephritis.
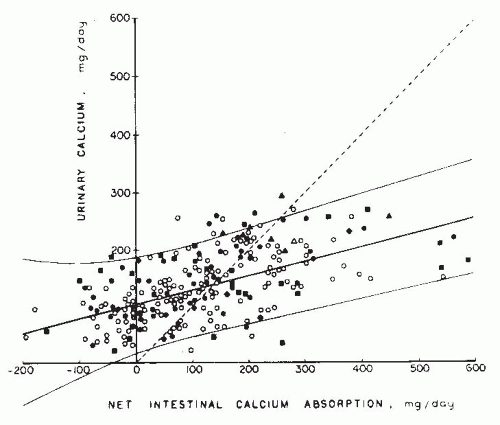 FIGURE 20.5 Urinary calcium excretion as a function of net intestinal calcium absorption. Data are derived from 6-day balance studies on 195 normal adults. Each symbol represents individual subjects from different sources: circles, Knapp238; squares, Lafferty and Pearson239; diamonds, Liberman et al.240; and triangles, Edwards and Hodgkinson.241 Open figures represent women, and solid figures represent men. Solid lines represent mean and 2 standard deviations. The dotted line is the line of identity; points above the line reflect negative calcium balance. (From Coe FL, Favus MJ. Nephrolithiasis. In: Brenner BM, Rector FC, eds. The Kidney, 2nd ed. Philadelphia: WB Saunders; 1991, with permission.) |
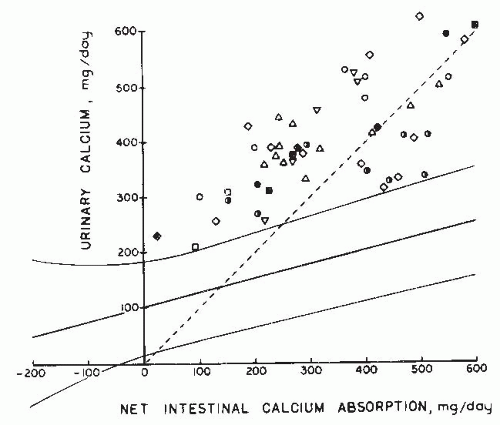 FIGURE 20.6 Urinary calcium excretion as a function of net intestinal calcium absorption from 6-day balance studies performed on 51 patients with idiopathic hypercalciuria reported as follows: open square, Henneman et al.243; open squares with dot in center, Jackson and Lancaster244; open triangles, Harrison245; open circles with dot in center, Dent et al.246; open inverted triangles, Parfitt et al.247; closed diamonds, Edwards and Hodgkinson241; open diamonds, Liberman et al.240; and half-darkened circles, Lemann.248 Solid lines represent mean and 2 standard deviations derived from balance studies from 195 normal adults, shown in Figure 26.10. The dotted line is the line of identity, with positive calcium balance below the line. (From Coe FL, Favus MJ. Nephrolithiasis. In: Brenner BM, Rector FC, eds. The Kidney, 2nd ed. Philadelphia: WB Saunders; 1991, with permission.) |
TABLE 20.3 Fraction of Filtered Calcium Excreted in the Urine by Normal and Hypercalciuric Subjectsa | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
above normal and increases the filtered load of calcium (Fig. 20.1). PTH secretion is reduced by the hypercalcemia, and suppression of PTH secretion would reduce calcium reabsorption because PTH stimulates renal tubular calcium reabsorption. In contrast, a renal tubular transport defect (Fig. 20.2) leading to hypercalciuria would produce secondary hyperparathyroidism. PTH, in turn, would stimulate the production of 1α,25(OH)2D3 and produce intestinal calcium hyperabsorption. Hyperabsorption would elevate postprandial serum calcium levels, raising the filtered calcium load and decreasing the magnitude of secondary hyperparathyroidism. The only way of distinguishing one mechanism from the other is by testing specific predictions that differ in the two forms of hypercalciuria. Clinically, PTH levels are the most clear-cut basis of distinction. Fasting hypercalciuria is not a means of detecting a renal calcium leak because it can be and is caused by exaggerated bone remodeling.
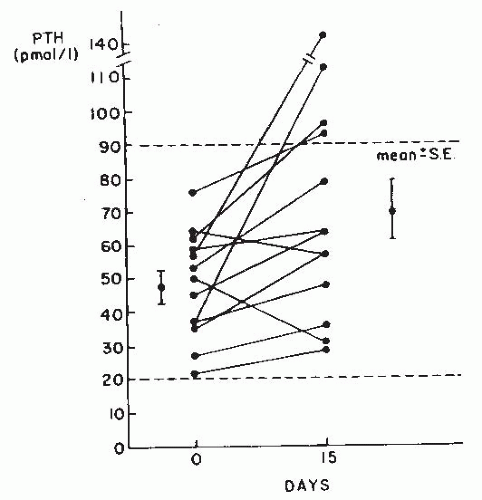 FIGURE 20.7 Changes in serum immunoreactive parathyroid hormone (PTH) in patients with fasting hypercalciuria after 15 days of diclofenac treatment. Dotted lines indicate the normal range of PTH. (From Filipponi P, Mannarelli C, Pacifici R, et al. Evidence for a prostaglandin-mediated bone resorption mechanism in subjects with fasting hypercalciuria. Calcif Tissue Int. 1988;43:61, with permission.) |
in the vitamin D receptor that have been incriminated in causing osteoporosis.112 Animal studies in the genetically hypercalciuric rat support the possibility that an abnormal vitamin D receptor could contribute to the pathogenesis of absorptive hypercalciuria.113
TABLE 20.4 Types of Renal Stones Formed and Frequency of Occurrencea | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|









