THYROID DISORDERS
Introduction
An appropriate supply of thyroid hormone is essential for normal metabolism, cardiovascular function, mental status, and muscle strength. Diagnosis of thyroid disease in patients on dialysis is complicated by the fact that kidney disease can produce many of the same findings as thyroid disease and is often caused or accompanied by diseases such as diabetes, connective tissue diseases, and liver disease. These conditions can confound physical findings or alter standard tests used to evaluate thyroid function. Thyroid physiology, the effects of kidney disease, the most common thyroid disorders, and their evaluation, and management are described in this section.
Thyroid Physiology
Thyroid function is regulated at multiple levels to ensure that only minimal variation in circulating thyroid hormone levels occurs. The complexity of regulation of thyroid hormone reflects its importance as a regulator of development as well as metabolism and essential systemic functions. The synthesis and secretion of thyroid hormone are primarily controlled by the hypothalamus and pituitary. Although T4 is the predominant circulating form of thyroid hormone, T3 is the biologically active form and is responsible for negative feedback at the levels of the hypothalamus and pituitary that can reduce thyrotropin-releasing hormone (TRH) and thyroid-stimulating hormone (TSH) secretion, respectively. The hypothalamus produces TRH, and TRH stimulates TSH production and release by the thyrotrophic cells in the anteromedial region of the pituitary. TRH production and secretion increase in response to reduced circulating levels of T3. TSH is the primary trophic hormone for the thyroid gland and is responsible for its size, vascularity, and the level of thyroid hormone production as well as the release of thyroid hormone.
Iodine, an essential substrate of thyroid hormone synthesis that is concentrated in the thyroid by an active transport mechanism, contributes to control of thyroid hormone synthesis and release. Excess iodine inhibits iodine uptake and thyroid hormone synthesis by the thyroid. High levels or acute high doses (such as given with Lugol’s solution or with iodinated contrast dye) reduce thyroid hormone synthesis and secretion. The primary route of iodine excretion is the kidney, and as renal insufficiency progresses, one would expect iodine to be retained. Dialysis patients have increased serum inorganic iodine and thyroid iodine content as well as an increased incidence of enlarged thyroid glands, but the full physiologic implications of this situation are not understood (1).
Both T4 and T3 are produced in the thyroid, but T4 is the predominant secreted form. T3 levels in the systemic circulation primarily reflect peripheral conversion of T4 to T3 by a monodeiodinase, a metabolic step that is also subject to regulation. Several proteins in the blood bind and transport thyroid hormone, including thyroid-binding globulin (TBG), prealbumin, and albumin. Except in rare circumstances, the effects of the other proteins are negligible compared to that of TBG. Loss of thyroid hormone through hemo- or peritoneal dialysis is negligible under normal circumstances (1). Finally, the sensitivity of tissues to thyroid hormone can be altered through changes in thyroid hormone receptor expression (2).
Serum thyroid hormone levels vary with changes in the concentration of serum thyroid hormone–binding proteins. In states such as liver disease (acute and chronic hepatitis, primary biliary cirrhosis), HIV infection, use of estrogen, tamoxifen, or pregnancy, TBG levels are increased, which will be reflected in increased total thyroid hormone. In the nephrotic syndrome, following administration of androgens, or high-dose glucocorticoids, and in major systemic illnesses or acromegaly, TBG levels are decreased, and total serum thyroid hormone levels are reduced. In the absence of other complicating factors, serum free T3 (fT3) and free T4 (fT4) levels, the biologically important parameters, are normal.
Peripheral conversion of T4 to T3 is controlled by at least three enzymes, types 1 (5′), 2 (5′), and 3 (5′) deiodinases. Type 1 deiodinase is expressed in liver, kidney, thyroid, the central nervous system (CNS), and the pituitary and is responsible for conversion of T4 to T3 in these tissues. This enzyme is primarily responsible for systemic T3 levels but may also produce reverse T3 (rT3), a metabolically inactive form of T3. The peripheral conversion of T4 to T3 (presumably through inhibition of this enzyme) is reduced in a number of conditions that are important in nephrology including renal failure, malnutrition, liver disease, other systemic illnesses, and following trauma or surgery (euthyroid sick syndrome, nonthyroidal illness syndrome, or NTIS). Drugs that impair conversion of T4 to T3 include glucocorticoids, propranolol (>200 mg/d), amiodarone, and oral cholecystographic agents. The type 2 deiodinase is expressed in the CNS, pituitary, brown fat, and placenta. In states (see subsequent text) where peripheral conversion of T4 to T3 is reduced, type 2 deiodinase may maintain the CNS, pituitary, brown fat, and placenta in a relatively euthyroid state through in situ production of T3. Type 3 deiodinase is expressed in the CNS, placenta, and skin and is the primary source of rT3. rT3 is an inactive metabolite of T4 that increases in some conditions including burns, trauma, and uremia. The fact that local concentrations of T3 may differ because of differential regulation of the deiodinases can make it difficult to determine if a hypothyroid state truly exists.
Measurement of Thyroid Function
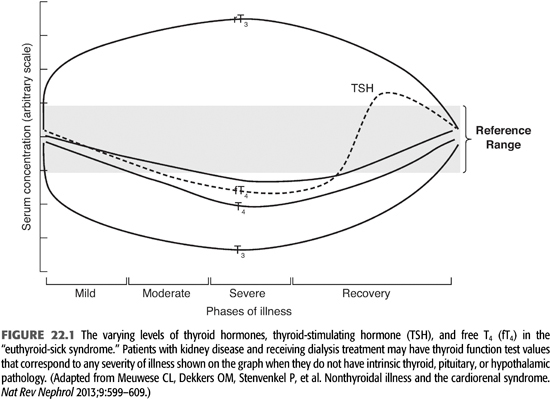
The suspicion of thyroid disease is raised by the patient’s history and physical findings and confirmed with laboratory measurement of thyroid hormone levels. Described below are the most commonly used assays with an emphasis on the laboratory characterization of the euthyroid-sick, NTISs because renal failure produces this pattern of laboratory values in up to 65% of dialysis patients (FIGURE 22.1 and TABLE 22.1) (3–5). Sensitive assays are available for measuring fT4, fT3, rT3, and TSH, and these measurements are the clinical tests used most commonly to establish the diagnosis of thyroid hormone excess or deficiency. Total serum T4, T3, and rT3 are measured by radioimmunoassay (RIA for rT3 is measured in special circumstances). Its level increases with alterations in peripheral deiodinase activity in systemic illnesses. T4 is tightly bound by serum proteins (primarily TBG), and the level of total T4 changes substantially with changes in the level of TBG. TBG may change markedly with disease or nutritional state so that a high or low total T4 level may not be meaningful if the level of the thyroid hormone–binding protein is also high or low. Renal failure and dialysis do not directly affect TBG levels, although its levels may be reduced with persistent nephrotic range proteinuria, high rates of peritoneal protein loss, or following androgen administration for anemia in patients who might not also be receiving erythropoietin.

The biologically active form of the hormone is that which is “free” in the serum and not protein bound. In the past two decades, improved assays have allowed for more accurate measurement of true free thyroid hormone levels. Thus, in states of “pure” thyroid disease, the lack of specificity for total T4 or T3 is no longer a diagnostic problem because the level of fT3 is a function of the level of fT4. However, in conditions such as kidney disease, malnutrition, or other systemic diseases (euthyroid sick or NTISs), the activity of the peripheral deiodinases is altered, and conversion of T4 to T3 is not regulated normally (5,6). In these situations, serum fT4 and fT3 measurement are essential to fully characterize thyroid function.
Sensitive assays exist that allow direct measurement of TSH, the trophic hormone from the pituitary that participates in feedback regulation of thyroid function. TSH levels are a function of T3 biologic activity in the hypothalamus where TRH production and release respond to T3 levels and T3 biologic activity in the pituitary. In primary thyroid disease, if T3 levels are reduced, TSH levels rise. If T3 levels are elevated, TSH levels are suppressed. However, hypothyroidism can occur as a result of a failure of the hypothalamopituitary axis resulting in low or inappropriately normal TSH levels, and thyrotoxicosis can result from increased secretion of TSH such as from a pituitary tumor. The normal range for TSH is 0.5 to 5 mU/mL. Patients who are thyrotoxic have serum TSH levels below 0.5 mU/mL, and patients who have hypothyroid have serum levels above 5 mU/mL. If pituitary failure is suspected, a TRH stimulation test can be performed.
Dialysis patients have at least one systemic illness, chronic kidney disease, and frequently others such as malnutrition and diabetes. These chronic illnesses (and others) alter the activity of peripheral deiodinases and cause an abnormal pattern of thyroid function tests, termed the euthyroid-sick syndrome (3,5). This patient population is also characterized by increased levels of circulating cytokines such as interleukin 6 (IL-6) that may also contribute to biochemical abnormalities including the euthyroid sick state (6). The euthyroid sick syndrome is characterized by reduced total and free serum T3 and increased serum rT3 as a result of increased peripheral deiodination of T4 to rT3. The degree of reduction of the serum T3 level corresponds to the severity of the systemic illness (FIGURE 22.1). In mild to moderately severe illnesses, fT3 is reduced, rT3 is increased, but T4, fT4, and TSH levels are normal. With severe illness, the fT4 levels may fall in the subnormal range. During recovery from a severe illness, the TSH level may rise to levels slightly above normal before normalizing. Consequently, thyroid hormone levels in dialysis patients must be interpreted using a euthyroid-sick pattern as normal.
Whether patients with the NTIS are in fact physiologically hypothyroid is not known. fT4, fT3, and rT3 levels reflect systemic metabolism of T4, while TSH levels reflect hypothalamic and pituitary levels of T3. In the presence of illness where peripheral conversion of T4 to T3 is reduced, TSH levels may not reflect low T3 levels because the conversion of T4 to T3 in the tissue responsible for sensing T3 activity (hypothalamus and pituitary) is mediated by a deiodinase whose activity does not appear to change with systemic illness. In contrast, systemic measurement of thyroid hormones reflects metabolism by different deiodinases whose activity may change with systemic illness. Consequently, the CNS and pituitary may appear euthyroid, while the remainder of the body is hypothyroid. Administration of L-T4 to severely ill patients has been studied but does not alter their outcomes (7,8). Survivors could be identified by their high baseline T3:T4 ratios. Low serum T3 levels in dialysis patients are associated with reduced survival (4). Preliminary studies in dialysis patients suggest that treatment of dialysis patients with low doses of T3 results in a less favorable nitrogen balance and increased protein degradation (1).
Thyroid Diseases
Thyroid diseases can be separated into those that cause increased hormone levels (thyrotoxicosis), decreased hormone levels (hypothyroidism), those that produce nonfunctional nodules, and the inflammatory diseases of the thyroid.
Thyrotoxicosis
Thyrotoxicosis is a pathophysiologic condition that results from excess thyroid hormone. The precise manifestations of thyrotoxicosis depend on the magnitude of hormone excess, the age of the patient, and the presence of other illnesses such as cardiovascular disease and diabetes. In general, the effects of thyroid hormone excess represent an exaggeration of its normal physiologic response and are usually most prominent on metabolism and on the cardiovascular and nervous systems.
Elevated thyroid hormone levels cause an increase in metabolic rate that is reflected in increased calorie utilization, oxygen consumption, heat generation, and increased basal body temperature. Protein synthesis and breakdown are increased, but degradation exceeds synthesis, leading to weight loss and decreased body mass. Many such patients have an abnormal glucose tolerance that may simulate diabetes mellitus. Lipid metabolism is also altered with both increased synthesis and degradation of lipids, but with degradation exceeding synthesis, leading to increased serum free fatty acid levels, while triglyceride and cholesterol levels are decreased. The metabolic manifestations of thyrotoxicosis may be difficult to detect in patients with renal disease and those who are on dialysis because they overlap with those of renal failure or with chronic diseases such as diabetes mellitus that are commonly found in the dialysis population. Although kidney disease patients rarely are hyperthermic, they commonly are catabolic and glucose intolerant.
Thyroid hormone has a direct cardiostimulatory activity that leads to increased cardiac output through resting tachycardia (heart rate greater than 100 bpm), increased stroke volume, and decreased peripheral resistance. The increased cardiac output is a response to supply the increased oxygen demands and the requirement to dissipate heat. Patients with hyperthyroidism may also have increased blood pressure and a widened pulse pressure. Because of the high frequency of hypertension and alterations in total body volume in dialysis patients, these findings may not be valuable in identifying patients with thyrotoxicosis. However, arrhythmias, congestive heart failure, and a change in pattern of angina may provide excellent clues to the presence of thyrotoxicosis. Supraventricular arrhythmias, especially atrial fibrillation, are common and may be the presenting complaint in patients with thyrotoxicosis. In the absence of preexisting heart disease, congestive heart failure is an uncommon presentation of thyrotoxicosis. Patients with thyrotoxicosis and atrial fibrillation are commonly resistant to heart rate control with digitalis. The frequency, pattern, or severity of angina is frequently increased in thyrotoxic patients with coronary artery disease. Patients may complain of shortness of breath in the absence of congestive heart failure. This sensation may be due to reduced lung compliance and muscle weakness in some circumstances.
Thyrotoxicosis usually produces prominent effects in the nervous system. Patients complain of nervousness that is characterized by a short attention span, restlessness, and compulsion to move despite fatigue. These patients often have fast jerky movements and a fine rhythmic tremor of hands and tongue. They are emotionally labile and often complain of insomnia. Psychiatric disorders occur including manic-depressive, schizoid, and paranoid states. In patients with seizure disorders, the seizure threshold is lowered resulting in an increased frequency of seizures. The hyperkinetic state described is uncommon in dialysis patients, but fatigue, restlessness, inattention, psychiatric and emotional disturbances, and changes in seizure frequency occur frequently. Consequently, the existence of any one of this constellation of findings in a patient may not be helpful in suggesting thyrotoxicosis, but their appearance in a patient who was previously stable may be of diagnostic value.
Common gastrointestinal (GI) symptoms include increased motility (faster gastric and intestinal emptying and transit times) with less well-formed stools, and increased stool frequency. Overt diarrhea is rare. Appetite is generally increased in hyperthyroid patients, although it may be decreased in severe cases or in the elderly. Patients with kidney disease usually have decreased bowel motility, so apparent normalization of bowel function may be a clue to thyrotoxicosis. Hepatic dysfunction with elevated transaminases has been reported in thyrotoxicosis, possibly due in part to increased O2 extraction in the splanchnic bed (9).
Increased levels of thyroid hormone alter the structure and function of muscle and bone. Patients complain of proximal muscle weakness and fatigability. These symptoms are usually partly due to wasting and loss of muscle mass but are also due to primary changes in the muscles. Myopathy is more common in men than women. In patients with normal kidney function, thyrotoxicosis leads to loss of bone mass with increased bone turnover, greater urinary excretion of Ca and PO4, and turnover markers such as deoxypyridinoline, with reduced levels of serum PTH and vitamin D. In dialysis patients, the effects of excess thyroid hormone on bone mass presumably are similar, but they have not been reported.
Causes of Thyrotoxicosis
Dialysis patients develop thyrotoxicosis for the same reasons as patients with normal kidney function. The causes include Graves’ disease or toxic diffuse goiter, toxic thyroid nodule, toxic multinodular goiter, thyroiditis, and rarely due to increased secretion of TSH due to a pituitary adenoma, increased secretion of TRH, or resistance of either the hypothalamus or pituitary to T3. Graves’ disease is the most common cause of thyrotoxicosis and is an autoimmune disease that results from circulating autoantibodies (thyroid-stimulating immunoglobulins or TSIs) that appear to be directed against the TSH receptor on the thyroid cell membrane. The disease is characterized not only by thyrotoxicosis but also by an infiltrative ophthalmopathy producing exophthalmos and dermopathy. Toxic multinodular goiter usually arises in a multinodular thyroid gland. The thyrotoxicosis is usually less severe than in Graves’ disease, but it occurs in an older population so cardiovascular symptoms may be more prominent (2). Thyroid nodules may be more common in patients receiving chronic dialysis (10). Thyroiditis may be triggered by a viral infection, exposure to iodinated contrast material, or medications such as amiodarone.
Diagnosis of Thyrotoxicosis
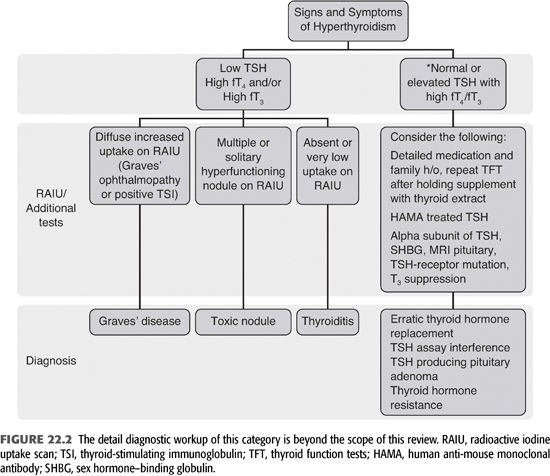
Thyrotoxicosis may not be suspected in dialysis patients because the symptoms of thyrotoxicosis may be ascribed to other underlying illnesses such as cardiovascular disease, hypertension, and volume overload. Many of the symptoms may be masked by medications such as β-blockers or other blood pressure medications. Weight loss may be masked by retained fluid or ascribed to gastroparesis. Nevertheless, new symptoms that are consistent with thyrotoxicosis, particularly cardiovascular symptoms, should suggest the possibility of thyrotoxicosis (TABLE 22.1 and FIGURE 22.2). The complaint or finding of increasing neck size or a lump in the anterior neck is particularly valuable in identifying patients who may be thyrotoxic. Thyrotoxicosis is caused by elevated T3, and a diagnosis of thyrotoxicosis requires elevated levels of fT3 and/or fT4, so these hormones should be measured. If thyrotoxicosis is caused by primary overproduction of thyroid hormone by the entire gland (Graves’ disease) or by nodules (95% of cases), the TSH level will be suppressed. If thyrotoxicosis is caused by overproduction of TSH (pituitary tumor, hypothalamic lesion or ectopic production, <5% of cases), the TSH level will be elevated.
Treatment of Thyrotoxicosis
The immediate treatment of thyrotoxicosis in patients with kidney disease is the same as other patients and includes the use of β-blockers, dexamethasone, and agents that block thyroid hormone synthesis and release (propylthiouracil and methimazole). In patients with renal failure, propylthiouracil can cause granulocytopenia, agranulocytosis, rheumatic syndromes, and hepatitis. However, the doses of both propylthiouracil and methimazole are not changed in dialysis patients. Radioactive 131iodine (131I) ablation may also be used, although it is cleared by both peritoneal and hemodialysis (11). Disposal of the radioactive effluent can be a problem for which guidelines do not exist.
Hypothyroid States
Hypothyroidism occurs for three general reasons: (a) most commonly, loss of functional thyroid tissue; (b) loss of trophic activity by TSH; and (c) defective hormone synthesis (as with iodine deficiency). Loss of trophic activity in pituitary or hypothalamic lesions represents approximately 5% of all cases of hypothyroidism. The symptoms of hypothyroidism are generally the opposite of those of thyrotoxicosis. They are even more difficult to identify in kidney disease and dialysis because like kidney disease, hypothyroidism results in generalized constitutional symptoms and gradual loss of function. Reduced glomerular filtration rate (GFR) is associated with hypothyroidism, and a GFR of <30 mL/min confers a risk of hypothyroidism that is increased approximately twofold over the normal population (GFR >90 mL/min) (12).
In hypothyroidism, the metabolic rate slows, resulting in decreased oxygen consumption; heat generation decreases; appetite decreases; and the rate of protein synthesis is reduced. The latter may be due to decreased effectiveness of GH and insulin-like growth factor 1 (also known as somatomedin C). The oral glucose tolerance test is flat, and the peak insulin response is delayed. The rate of degradation of insulin is decreased leading to an increased sensitivity to exogenous insulin. Patients also have increased capillary permeability which leads to peripheral edema. Cholesterol and triglyceride circulating concentration levels are elevated as a consequence of their decreased clearance, and high-density lipoprotein (HDL) levels are reduced. Superficially, none of these findings are specific for hypothyroidism and are seen frequently in dialysis patients.
Hypothyroidism reduces heart rate and myocardial contractility, diminishing pulse pressure and reducing cardiac output. The heart is enlarged, and a pericardial effusion frequently develops. Pleural effusions are also common. Electrocardiogram (ECG) abnormalities include sinus bradycardia, prolonged PR interval, reduced amplitude of the P wave and QRS complex, and nonspecific ST-segment and T-wave changes. These changes may contribute to congestive heart failure. Other than the bradycardia, these findings are commonly seen to progress slowly over months and are commonly seen on dialysis services in patients with normal thyroid function.
In hypothyroidism, GI motility is decreased and constipation is a frequent complaint. The mucosa of the GI tract is atrophic causing reduced absorption, but the decreased transit time partially compensates for the decreased absorption rate. Ascites consisting of mucopolysaccharide- and protein-rich fluid is common. Superficially, these findings are not pathognomonic for hypothyroidism and can occur for a variety of reasons in the dialysis population.
The most prominent symptoms in hypothyroidism are frequently found in the nervous system. They include reduced cognitive function, deterioration of memory, slow speech, syncope, and coma. Psychiatric symptoms may include paranoia or depression. Speech is thick and slurred as a consequence of infiltration of the larynx and tongue by mucopolysaccharides. Movements are slow and appear uncoordinated and may be further compromised by compression of peripheral nerves by myxedematous deposits. Reflexes are slow due to altered nerve responses including delayed relaxation time. Myxedematous patients complain of stiff, sore muscles, but muscle strength usually remains intact.
Causes of Hypothyroidism
Loss of functional thyroid tissue occurs following ablation of hyperactive thyroid tissue (such as following ablative therapy for Graves’ disease with 131I), idiopathic hypothyroidism, or in the late stages of Hashimoto’s thyroiditis (autoimmune thyroiditis). Excess iodine inhibits thyroid function by inhibiting thyroid uptake of iodine and thyroid hormone synthesis and release. As the primary route of excretion of iodine is the urine, one might expect high levels of iodine to accumulate in patients with renal failure. Regulation of iodine metabolism in kidney disease and dialysis patients has not been studied systematically, but 131I is removed by hemodialysis and peritoneal dialysis (11). One report of three cases exists of hypothyroidism due to iodine accumulation in dialysis patients eating a high-iodine diet (13). Iodinated contrast agents may also represent an iodine load for dialysis patients that could affect thyroid function. Another group of patients who may be at risk for hypothyroidism are patients with iron overload syndromes (14). Metabolic acidosis causes reduced T3, and T4, levels and increased TSH levels with an increased sensitivity of TSH secretion to TRH stimulation (15). In dialysis patients, correction of metabolic acidosis increases TSH levels so the metabolic acidosis associated with kidney disease is a treatable cause of hypothyroidism (16).
Diagnosis of Hypothyroidism
Many of the symptoms of hypothyroidism are nonspecific and overlap with those of chronic kidney disease (17). Consequently, hypothyroidism may not be suspected and may go undiagnosed. Patients who are at particular risk for hypothyroidism are those who have a history of previous ablative treatment of thyroid disease, possibly those with other autoimmune endocrine diseases, or those whose general condition deteriorates for unexplained reasons. The physical signs of hypothyroidism such as delayed reflexes may be undetectable in a diabetic patient with neuropathy, or a slow pulse may be masked in a patient taking β-blockers, and may also be difficult to identify in dialysis patients who have many comorbid conditions and take many medications. Nevertheless, if present, delayed reflexes are valuable in making the diagnosis of hypothyroidism as long as other neurologic disorders are not present. If a patient is not taking medications that could slow the heart rate, then heart rate is also valuable in screening patients for hypothyroidism. Measurement of circulating thyroid hormone levels will usually reveal a “sick-euthyroid” pattern with a variable (low or normal) fT4, low rT3U, low fT3, and normal or mildly elevated TSH. However, dialysis patients who are truly hypothyroid will have elevations of TSH with normal hypothalamopituitary function, so primary hypothyroidism can be diagnosed with a serum TSH in most situations. Whether mild elevations of TSH represent subclinical hypothyroidism, or whether this state represents a euthyroid CNS and pituitary with a hypothyroid periphery is not clear (2). Normal patients with minimal elevations of their TSH levels (<10 to 15 mU/mL) are usually not symptomatic. Isolated hypothyroidism is unusual in pituitary or hypothalamic lesions (see subsequent text), but if hypothyroidism is due to pituitary or hypothalamic failure (tumors, following radiation therapy, trauma), the TSH level may be extremely low (less than 0.5 mU/mL). In these situations, evaluation of the pituitary and hypothalamus structure with either computed tomography (CT) or magnetic resonance imaging (MRI) scans and function with TSH and TRH stimulation tests is indicated. Corroborative evidence of hypothalamic or pituitary disease with reduced follicle-stimulating hormone (FSH), luteinizing hormone (LH), GH, and cortisol levels should also be sought.
Treatment of Hypothyroidism
Thyroid replacement therapy in hypothyroid patients on chronic dialysis is the same as that in other populations. In general, patients with kidney disease are at risk for cardiovascular disease if they have not already been diagnosed with it. Consequently, their thyroid hormone should be replaced slowly to avoid precipitating angina or congestive heart failure, unless the patient is comatose or severely ill with another diagnosis. The usual replacement dose of levothyroxine (T4) is around 1.6 μg per kilogram body weight per day. Some physicians use thyroid extract (120 mg/d) or T3 (liothyronine or cytomel, replacement dose 50 to 100 μg/d), but levothyroxine (T4) has the advantage that the body can regulate its conversion to T3 and the serum half-life is longer (18). A role for T3 in euthyroid-sick patients has not been established (2). The effectiveness and dosing of thyroid replacement therapy is usually followed by measuring serum TSH because no other physiologic or symptomatic parameter is sufficiently sensitive.
 ANTERIOR PITUITARY DISORDERS
ANTERIOR PITUITARY DISORDERS
Introduction
The anterior pituitary gland integrates information from the brain (hypothalamus) and feedback from peripheral hormones to control production of its trophic hormones (19,20). The hormones produced by the anterior pituitary, adrenocorticotropic hormone (ACTH), GH, prolactin, TSH, FSH, and LH, all have specific effects on different tissues in the body, and their excess production as well as loss of their production produces specific syndromes. In general, syndromes of pituitary hormone excess can be related to individual hormones because they are individually controlled and produced in distinct cell types. In contrast, deficiency syndromes involve multiple or all of the hormones secreted because they are caused by processes that destroy the hypothalamus and/or pituitary. Prolactin is the exception because its synthesis and release are under tonic inhibitory control by the hypothalamus. Chronic kidney disease alters the regulation and response to GH, prolactin, FSH, and LH. The biology of these hormones is complex and not fully understood in normal conditions, and their biology remains only partially characterized in dialysis patients. TSH is discussed earlier in the section “Thyroid Diseases.”
Physiology
The anterior pituitary contains cells that produce several hormones including ACTH, GH, prolactin, TSH, FSH, and LH. Each hormone is produced by an individual cell type. Functioning tumors of the pituitary are usually clonal and produce one hormone of the anterior pituitary. The venous system that drains the hypothalamus forms a plexus in the anterior pituitary so that peptide-releasing factors descending from the hypothalamus control the activity of the pituitary cells and the release of ACTH, GH, prolactin, TSH, FSH, and LH. The six peptide-releasing factors are corticotrophin-releasing factor (CRF) that stimulates ACTH release, TRH that stimulates TSH release, gonadotropin-releasing hormone (GnRH) that simulates FSH and LH release, growth hormone–releasing hormone (GHRH) that stimulate GH release, somatostatin that inhibits secretion of all of the pituitary hormones but is most potent in inhibiting GH release, and prolactin inhibitory activity (PRIH) that tonically inhibits prolactin release. PRIH may be several factors including dopamine. These hormones are also subject to regulation at the level of the pituitary by direct feedback mechanisms.
Growth Hormone
GH has a variety of growth-promoting and metabolic effects (19,20). GH is produced in the somatotrophs of the pituitary and is secreted under the dual control of GHRH (stimulatory) and somatostatin (inhibitory), which results in a pulsatile pattern of release. GHRH secretion is increased by dopamine, serotonin, α-adrenergic agonists, hypoglycemia, exercise, protein-rich food, and emotional stress and is inhibited by β-adrenergic agonists, free fatty acids, insulin-like growth factor 1, and GH. GH is carried in the serum primarily by a binding protein, growth hormone–binding protein (GHBP), which is derived from the GH receptor. Although GH has direct differentiating effects on some cell types, most of its effects are through stimulation of production of somatomedins or insulin-like growth factors 1 and 2 (IGF-1 and IGF-2). IGF-1 appears to replicate many of the biologic effects of GH, but the function of IGF-2 is less well defined. IGF-1 is produced locally in small quantities in response to GH, but the liver appears to be the main site of production. IGF is carried in the serum by a six-member family of binding proteins (IGFBP 1–6) and is so tightly bound that only approximately 1% of the hormone is free.
The growth-promoting effects of GH are primarily to increase cell number and produce a positive nitrogen balance, with accumulation of Ca, Mg, K, Na, and PO4. Before puberty and the closure or epiphyses, GH increases bone length and thickness and produces a proportional increase in the size of other organs and tissues except the CNS. In adulthood (after the closure of epiphyses), bones cannot grow in length and only become thicker while other organs increase in size. Like insulin, GH is an anabolic hormone, but in contrast to insulin, it leads to carbohydrate intolerance, inhibits lipogenesis, increases mobilization of fat, and promotes ketosis. In nondiabetic patients with even low amounts of insulin present, these effects of GH are anabolic, but in diabetic patients, its effects are diabetogenic. In the absence of insulin, GH does not have anabolic activity. GH levels increase with exercise, hypoglycemia, and sleep.
GH excess causes gigantism if it occurs before puberty, and acromegaly if it occurs after epiphyseal closure. The most common cause of GH excess is a microadenoma of the pituitary. GH deficiency may occur as a consequence of hypothalamic disease or as a result of loss of GH receptors (Laron dwarfism). If it occurs in infancy or childhood, growth failure results. In adults who are fully grown at the time GH deficiency develops, the consequences appear to be minimal and may involve mild loss of muscle mass and impaired nutrient utilization, but the condition has not been studied well.
Chronic kidney disease and dialysis alter the GHRH-GH-IGF-1 system such that uremic patients appear to be insensitive to GH and IGF-1 and have higher GH levels due to increased secretory bursts and an increased half-life of GH with reduced IGF-1 levels (16,21–25). This resistance to GH and IGF-1 may explain why acromegaly is so rare in dialysis patients. This field is complicated because the nutritional and metabolic status (metabolic acidosis) of the patient and chronic illnesses alter the GH/IGF axis (16). Consequently, the relative importance of malnutrition, acidosis, uremic toxins, and decreased clearance of peptides by the kidney are difficult to determine. This remains an area of intense investigation in both clinical and basic science. Most of the recent information is from the pediatric literature because of interest in the growth retardation found in pediatric renal failure and dialysis patients. In the adult population, insensitivity to GH and IGF-1 may contribute to the catabolism and wasting that commonly occur. Information on the mechanisms of resistance to GH and IGF-1 has not been completely established in humans. Some information is available from human studies and more can be inferred from animal disease models.
In kidney disease, GH levels are increased due to reduced clearance of GH by the kidney and an increased number of secretory bursts. In one study, the circulating concentrations in controls were 0.7 μg/mL versus 1.22 μg/mL in dialysis patients (21). In prepubertal patients, secretion is increased, but in pubertal patients and presumably adults, it is decreased (23,26). The level of GHBP (which reflects hepatic GH receptor expression levels) is reduced, providing a possible mechanism for GH insensitivity. Studies in humans and experimental animals indicate that metabolic acidosis is sufficient to confer resistance to GH (16,24,27,28). Dialysis patients are resistant to the short-term metabolic effects of IGF-1, IGF-1-stimulated reductions in plasma insulin, cortisol, C-peptide, and amino acid levels (29). Although the levels of IGF-1 are normal to slightly reduced, the levels of IGFBP (particularly IGFBP-2, 3, and 5) are increased (30). Because these IGFBPs bind IGF-1 tightly, the bioavailability of IGF-1 is reduced (23). The levels of the IGFBPs appear to be increased because of their reduced clearance by the kidney. In experimental systems, excess IGFBP is sufficient to inhibit the growth-promoting effects of IGF-1 (23). The half-life (t1/2) and volume of distribution of recombinant human IGF-1 are reduced in dialysis patients (31). Low molecular weight substances in uremic serum also appear to inhibit the action of IGF-1 (23,32).
The biologic effects of GH and IGF-1 are inhibited at many levels in uremia, and tissue resistance to these hormones may contribute to morbidity and mortality in this patient population. The benefits of human growth hormone (hGH) therapy are most clear in the pediatric population, and recombinant human growth hormone (rhGH) treatment is now established therapy for children with growth failure due to ESKD and renal insufficiency once nutritional and metabolic deficiencies (including acidosis, PO4 levels, and PTH levels) have been corrected or optimized (33). Long-term (4 to 8 years) administration of recombinant hGH (achieving supranormal levels and increasing IGF-1 levels) to children with chronic renal insufficiency and failure can restore linear growth and result in catch-up growth so that they achieve normal height (34,35). rhGH treatment may also improve muscle mass and bone density. This therapy does not have adverse effects on Ca, PO4, bone metabolism, glucose tolerance (although insulin levels may be elevated), the incidence of malignancy, or benign intracranial hypertension, and for patients with kidney insufficiency, the rate of progression of kidney disease was not accelerated (33–35). The dose of rhGH is higher than for patients with GH deficiency, on the order of 0.35 mg/kg/wk (28 IU/m2/wk), and may be higher during puberty. Nutritional requirements may increase in patients treated with rhGH. Methods for following dosing of the drug (e.g., serum IGF-1 levels) have not been established. Factors that contribute to a favorable response to hGH are increased height at the initiation of therapy, young age at the start of therapy, and shorter duration of dialysis treatment (33,35).
The role for recombinant hGH in the treatment of adults on dialysis is less clear because long-term, large-scale studies have not been performed. Patients treated with rhGH for 6 months show an increase in muscle mass, hand grip strength, albumin levels, and increased bone turnover with a decrease in bone density and total body mineral content (36–40). Shorter term metabolic studies demonstrate that rhGH treatment leads to a positive nitrogen balance (40). Although these studies support an anabolic effect of rhGH in dialysis patients and do not report complications of therapy, they are of relatively short duration, so the risk of potential complications of elevated levels of GH and IGF-1 that include insulin resistance and glucose intolerance, hypertension, cardiomyopathy, abnormal bone growth, and an increased incidence of colonic polyps cannot be determined. Optimal treatment of acidosis may restore the sensitivity of patients to GH and IGF-1, so improvements in the GH-IGF-1 system may be effected without administration of rhGH (41,42). Erythropoietin may sensitize the pituitary to the effects of GHRH, so optimal therapy with this agent may also have anabolic effects (43,44). The leptin system does not appear to play a major role in the catabolic state of dialysis patients (45,46).
Prolactin
Prolactin is produced by the lactotrophs in the pituitary, and it is secreted in a pulsatile fashion. In contrast to other pituitary hormones, prolactin is under tonic negative control by inhibitory factors from the hypothalamus. Its normal function in women is to promote lactation in cooperation with estrogen and progesterone, but its function in men is not established. Increased prolactin levels are caused by dopamine antagonists, oral contraceptives, diseases of the hypothalamus or pituitary that interfere with tonic suppression of prolactin secretion, and tumors. Up to 30% of pituitary tumors secrete prolactin. Prolactin is a stress response hormone, and its blood levels may rise with stress or pain. Normal prolactin levels are 2 to 15 μg/mL, levels in dialysis patients are often in the range of 15 to 50 μg/mL, and levels greater than 250 μg/mL are suggestive of first trimester pregnancy or a prolactin-secreting tumor (20,21). In women, excess production of prolactin results in galactorrhea, suppression of menstrual cycles, and infertility, and in men, infertility (47).
In dialysis patients, prolactin levels are elevated because of increased secretion and decreased clearance (21,47). Elevated prolactin levels may be responsible for some of the gonadal abnormalities found in kidney disease. Recent reports indicate that erythropoietin reduces prolactin levels by mechanisms that may be independent of the improvement in anemia (48). Hypothyroidism, pregnancy, and medication-induced hyperprolactinemia should be excluded by careful history, physical exam, TSH, and human chorionic gonadotropin (hCG) measurements. Prolactinomas can be effectively treated with dopamine agonist that suppresses prolactin production. Sexual function in dialysis patients has improved in response to bromocriptine, a dopamine agonist that suppresses prolactin production (49). Cabergoline is more effective than bromocriptine because cabergoline is better tolerated and has higher affinity for dopamine receptor binding sites (50,51). However, about 10% to 15% of patients with prolactinomas are resistant to dopamine agonist therapy and may require additional therapy such as transsphenoidal surgery or radiotherapy (52).
Follicle-Stimulating Hormone, Luteinizing Hormone, and Gonadal Function
FSH and LH are secreted by the same pituitary cell, the gonadotrope. Their secretion is controlled by the integration of signals from luteinizing hormone–releasing hormone (LHRH) and feedback signals from estrogen, progesterone, and androgens, and gonadal peptides such as inhibin (20). Pulsatile, rather than continuously elevated, levels of LHRH are required for normal gonadal function. The frequency and amplitude of the pulses is controlled by the hypothalamus through hypothalamic function and feedback mechanisms from the periphery. LH controls production of estrogen and progesterone in the ovary and testosterone in the testis. FSH stimulates sertoli cells to produce sperm and expression of LH receptors by Leydig cell. In women, FSH stimulates follicle development. These systems are abnormal in both men and women with kidney disease or who are on dialysis. Although psychological factors may contribute to sexual dysfunction, abnormalities at the levels of the hypothalamus and gonads are probably the predominant cause.
Reproduction in Women
Female dialysis patients frequently exhibit amenorrhea, dysmenorrhea, dysfunctional uterine bleeding, and cystic ovarian disease. These disorders occur because of disruption of normal menstrual cycles at the level of the hypothalamus and ovary (47,53). LH levels are tonically elevated due to increased release, the estrogen-induced LH surge does not occur, and FSH levels are normal or slightly elevated (53). Suppression of pulsatile LHRH secretion by prolactin may contribute to this problem. Additionally, the ovaries have a subnormal steroidogenic response to LH. The frequency of menstruation has risen from 10% to 20% 20 years ago to approximately 40% at this time which may reflect improved dialysis quality or care, but these cycles are generally thought to be anovulatory (54).
Conception occurs with a frequency of approximately 0.3% to 2.2% per year in Western countries, although these numbers represent estimates because accurate records are not kept (54–58). The frequency of conception on dialysis appears to be increasing, which may reflect improved dialysis techniques, beneficial effects of erythropoietin, and in some cases the effects of frequent dialysis regimens. Of the patients who conceive and progress to the first trimester, approximately 50% deliver viable infants. In two studies of patients who were treated with dialysis when they were pregnant, those who conceived before starting dialysis delivered viable infants in 75% to 80% of the cases, and those who conceived on dialysis delivered viable infants in 40% to 50% of cases (55,58,59). Pregnancies are complicated by intrauterine death, hypertension, intrauterine growth retardation, premature labor, premature birth of babies with low birth weights for gestational age, and an increased frequency of congenital malformations (60). Neonatal care is an important factor in the survival of these infants (55–58). Deliveries of viable infants occur at approximately 32 weeks, and the outcome improves with longer gestation. The infants weigh on the order of 1,200 to 1,550 g at birth. An increased dose of dialysis appears to be beneficial with reports of weekly Kt/V values of 6 to 8, or dialysis 5 to 6 days per week (55–57,61,62). Increased doses of erythropoietin are required to maintain hemoglobin levels in an acceptable range, and transfusions are sometimes required in addition (55,58). In many cases, patients are hospitalized around week 20 of gestation for management of blood pressure, dialysis fluid balance, nutrition, and anemia (63). Transplantation provides the best outcomes in fertility and pregnancy for patients with ESKD.
Evaluation for amenorrhea, dysmenorrhea, or dysfunctional uterine bleeding in women receiving dialysis should be carried out in consultation with a gynecologist. The evaluation should include a thorough pelvic examination, Pap smear, and may also include measurement of serum prolactin, LH and FSH levels, a pelvic ultrasound, and endometrial biopsy.
Reproduction in Men
In men, erectile dysfunction, impotence, decreased libido, and decreased sperm count are manifestations of uremia. LH is elevated due to increased secretion and decreased clearance. The increased LH production may be caused by low levels of testosterone synthesis by the testes. FSH levels are usually normal but may be high with severe testicular dysfunction. Presumably, increased prolactin levels would interfere with normal regulation of LHRH secretion. Evaluation of men for impotence should include measurement of serum prolactin, testosterone, FSH, and LH levels. Correction of anemia with erythropoietin may improve the function of the hypothalamo–pituitary–testicular axis in dialysis patients (64). Patients should also be evaluated for autonomic neuropathy and peripheral vascular disease since these two entities are relatively common in dialysis patients, especially those with hypertension and diabetes. Following kidney transplantation in young men, testicular size, sperm counts, and fertility commonly remain abnormal. These findings may reflect effects of antirejection regimens but nevertheless are a consequence of ESKD (65).
In contrast to women whose primary reproductive endocrinologic defect appears to be hypothalamic, hypogonadism is more prominent in men (66). These men, who have reduced testosterone levels, may respond to replacement testosterone. Erectile dysfunction is common in dialysis patients, affecting 70% to 80% of men (67). The incidence is higher in men with diabetes and who have been on dialysis for long periods, and these men tend to have reduced testosterone levels and reduced penile blood flow. Approximately 80% of men with erectile dysfunction respond to sildenafil (Viagra) (68). The level of penile blood flow is the most important parameter for predicting a response to sildenafil.
Adrenocorticotropic Hormone
ACTH stimulates steroidogenesis in the glomerulosa and fasciculata layers of the adrenal glands. ACTH is derived from proopiomelanocortin (POMC), a peptide that is produced by the corticotrope cells of the anterior pituitary and cleaved to ACTH and β-lipotropin (LPH). ACTH is further cleaved into α-melanocyte-stimulating hormone (α-MSH or ACTH 1-13) and ACTH-like peptide (ACTH 18-39). β-Lipotropin is processed to lipotropin (LPH) and β-endorphin. Secretion of ACTH is stimulated by corticotropin-releasing hormone (CRH) and vasopressin. Negative feedback control is provided by suppression of CRH, vasopressin, and ACTH secretion by cortisol, and inhibition of CRH secretion by ACTH. ACTH is secreted in a pulsatile fashion in a diurnal pattern that is maximal in the early morning and minimal in the evening.
In dialysis patients, ACTH levels are normal or slightly increased compared to normal controls, and its metabolism does not appear to be altered. Normal and abnormal responses to dexamethasone and metyrapone suppression tests have been reported. The circadian rhythms and levels of cortisol are normal, and the response of the adrenals to ACTH stimulation is normal. Consequently, a number of the tests of pituitary–adrenal function used in normal patients are applicable to dialysis patients.
 DISORDERS OF THE ADRENAL CORTEX
DISORDERS OF THE ADRENAL CORTEX
Introduction
The adrenal cortex produces two physiologically important steroid hormones, cortisol and aldosterone. The primary physiologic effect of aldosterone is on the kidneys, where it acts on the distal nephron to promote Na reabsorption and K and H excretion. Aldosterone also affects transport in the colon, but this appears to be a minor effect, and disorders of aldosterone metabolism in dialysis patients have not been reported. Consequently, this section deals with glucocorticoid excess or insufficiency.
Glucocorticoids are essential for life and affect glucose and lipid metabolism, the immune system, and bone and mineral metabolism. No tissue or organ system including the cardiovascular, GI, or CNS functions normally in the absence of glucocorticoids. Glucocorticoid activity is required for glycogen synthesis and maintenance of glycogen stores, gluconeogenesis, and has a permissive effect on lipolysis. The effects of glucocorticoids on the immune system under normal physiologic conditions are not fully understood, but pharmacologic concentrations decrease the number of peripheral lymphocytes, inhibit T-cell activation, macrophage proliferation, and inhibit the actions of a number of mediators of inflammation including chemokines, prostaglandins, and histamine. Glucocorticoids affect bone formation by reducing osteoblast numbers, decreasing intestinal Ca reabsorption, and increasing serum PTH levels.
Adrenal Insufficiency
In the general population, primary adrenal insufficiency usually presents as adrenal crisis following some form of stress such as surgery or an acute illness. Patients complain of anorexia, nausea, vomiting, and weakness with a history of weight loss. The findings may include volume depletion, hypotension and shock, hypoglycemia, fever, and possibly hyperpigmentation. This constellation of findings is caused by loss of both cortisol and aldosterone. The loss of aldosterone is largely responsible for the symptoms and findings related to volume depletion. Patients who are receiving renal replacement therapy but who encounter a physiologic stress can also develop adrenal crisis (25,69).
Dialysis patients have minimal renal function and therefore do not develop the salt-losing nephropathy associated with adrenal insufficiency. Presumably, if dialysis patients develop adrenal insufficiency, they present more like patients with secondary adrenal insufficiency (pituitary lesions with loss of ACTH production) or tertiary (hypothalamic lesions with loss of CRF production). In these patients, adrenal crisis is rare, and they present with insidious onset of generalized symptoms of malaise and signs and symptoms of hypoglycemia.
Primary adrenal insufficiency is caused by tuberculosis, fungal diseases, metastases from tumors, and autoimmune disease. As dialysis patients are at increased risk for tuberculosis, they are also presumably at increased risk for adrenal insufficiency due to tuberculosis. Autoimmune adrenalitis is associated with polyglandular autoimmune syndromes I and II which are associated with mucocutaneous candidiasis and hypoparathyroidism, and thyroid disorders and insulin-dependent diabetes, respectively. Primary autoimmune polyglandular syndrome type II is more common than the autosomal recessive type I syndrome. Patients with HIV infections and with cytomegalovirus, mycobacterium avium intracellulare, cryptococcus, or Kaposi’s sarcoma may manifest partial adrenal insufficiency. Adrenal insufficiency can be precipitated by ketoconazole because it inhibits cortisol synthesis, or rifampin because it increases cortisol metabolism (69). Secondary or tertiary adrenal insufficiency is associated with destructive lesions of the pituitary or hypothalamus such as tumors, postradiation therapy, trauma, or use of anticoagulants. In these situations, other pituitary hormones are usually lost in addition to ACTH. Adrenal insufficiency is also caused by long-term use of glucocorticoids for immune suppression as in patients with systemic lupus erythematosus, other glomerular diseases, or following renal transplantation.
Diagnosis of Adrenal Insufficiency
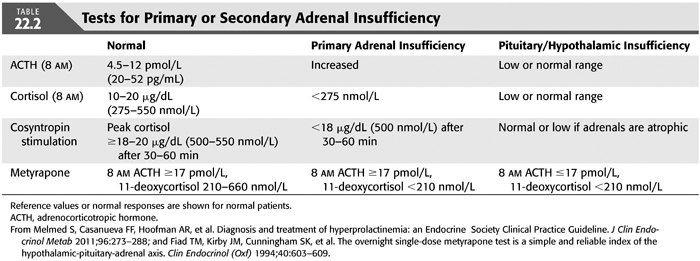
Renal failure and dialysis do not alter the diurnal variation of cortisol or the adrenal response to ACTH (25). Consequently, AM and PM cortisol levels and the ACTH stimulation test can be used to demonstrate adrenal or hypothalamic–pituitary–adrenal function (TABLE 22.2 and FIGURE 22.3). As serum cortisol concentrations are normally highest in the early morning hours ranging between 10 and 20 μg/dL (275 to 555 nmol/L), inappropriately low cortisol less than 3.6 μg/dL (100 nmol/L) strongly suggest adrenal insufficiency (70). Values below 10 μg/dL (275 nmol/L) make the diagnosis likely but require additional standard or low-dose ACTH stimulation test (cosyntropin test). In the standard ACTH stimulation test, plasma cortisol is measured before, 30 minutes after, and 60 minutes after intravenous injection of 250 μg of synthetic ACTH (ACTH 1-24). A cortisol value ≥18 to 20 μg/dL (500 to 550 nmol/L) at any of the time points in the test indicates normal adrenal function. The low-dose ACTH stimulation test is preferred and has improved sensitivity in cases of secondary adrenal insufficiency of recent onset or partial adrenal insufficiency. The prolonged ACTH stimulation test or an insulin-induced hypoglycemia test helps differentiate between primary and secondary or tertiary adrenal insufficiency (71–73). The overnight, single-dose metyrapone suppression test with measurement of serum cortisol, 11-deoxycortisol, and ACTH is useful for making a diagnosis of complete or partial pituitary-adrenal insufficiency and is the most sensitive test of pituitary-adrenal reserve (69). Patients may have a normal response of ACTH and cortisol to hypoglycemia but have an abnormal response to metyrapone suppression. Metyrapone blocks conversion of 11-deoxycortisol, a compound with no glucocorticoid activity and that does not participate in feedback to cortisol. As cortisol production decreases, ACTH rises and stimulates the production of cortisol precursors which can be measured as 11-deoxycortisol. Metyrapone (30 mg/kg body weight) is given orally at midnight, and 8 AM cortisol, 11-deoxycortisol, and ACTH levels are measured. A normal response is a rise in 11-deoxycortisol from 210 to 660 nmol and a level of ACTH greater than 17 pmol/L. If the cortisol level is normal or greater than 210 nmol/L in the 8-AM sample, the dose of metyrapone was inadequate to block cortisol synthesis. A normal response to the metyrapone suppression test excludes abnormalities of the hypothalamo–pituitary–adrenal axis. ACTH release can also be measured in response to CRF to document intact pituitary function (69). Patients who are addisonian may become symptomatic with the metyrapone suppression test, so it should be supervised. If primary adrenal insufficiency is suspected, a CT scan of the abdomen to evaluate the size of the adrenal glands is warranted.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


