Fig. 20.1
Light chain deposition disease. Glomeruli showing (A) PAS and (B) silver positive mesangial nodules. Note the lobular accentuation of the glomerular tufts. Arrows point to mesangial nodules. (PAS- periodic acid Schiff stain, Silver- Jones methanamine silver stain, both 40x). Courtesy Dr. Sanjeev Sethi, Mayo Clinic
Immunofluorescence is the most sensitive technique to diagnose MIDD on renal biopsy and is essential for diagnosis [2, 6, 9]. The light chains present in LCDD are more commonly kappa than lambda, with the occurrence of lambda light chains only accounting for 10–30 % of cases [2, 8, 13, 17]. A recent case series by Nasr et al. of 64 patients with MIDD showed that 84 % of LCDD patients and 67 % of LHCDD patients had a kappa light chain present on immunofluorescence [9]. In addition, kappa light chain nephropathy is more likely to be associated with nodular glomerulosclerosis than lambda light chain nephropathy [7]. The light chains show a predilection for the basement membranes in the tubules and the glomeruli [5] (Fig. 20.2). The most common site of basement membrane deposition is the tubule, followed by the glomerulus and the blood vessel [6]. Electron microscopy reveals granular, “powdery,” electron-dense deposits along outer aspect of the tubular basement membrane and the inner side of the glomerular basement membrane, as well as in the mesangium (Fig. 20.3) [12].
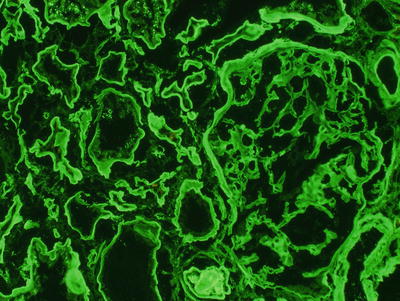
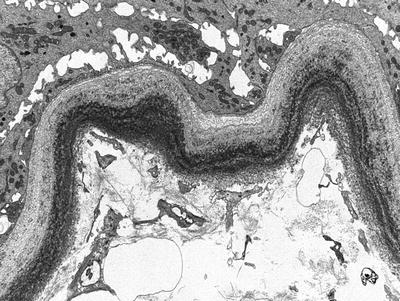
Fig. 20.2
By immunofluorescence, a stain shows bright linear glomerular and tubular basement membrane staining for kappa light chain; staining for lambda light chain and other immunoreactants was negative
Fig. 20.3
Electron micrograph of a tubule shows finely granular electron-dense deposits along the tubular basement membrane. Similar deposits were seen along glomerular basement membranes and in the mesangium
The heavy chain in both LHCDD and HCDD is more likely to be of the γ[gamma] subtype, and immunofluorescence reveals IgG staining that is monotypic on IgG subclass staining. The staining pattern and deposits on electron microscopy are otherwise indistinguishable from LCDD. All IgG subclasses (1–4) have been reported and there are rare cases of IgA-HCDD [18].
There have been uncommon cases of MIDD in which electron-dense deposits were demonstrated on electron microscopy despite negative immunofluorescence. This discrepancy is more common in patients with coincident myeloma cast nephropathy (MCN) [2, 11].
As immunoglobulins are capable of causing several different disease processes, it is not surprising that features of other paraprotein-associated diseases can frequently be seen on renal biopsy. LCDD may co-occur with AH, AL, and MCN. Pozzi et al. reported a case series of 63 patients in which 3 % had amyloidosis on renal biopsy [2]. In one small case series, all three patients had renal biopsies with features consistent with both LCDD and AL [19]. Other tissues that were sampled in two of these patients—including skin, liver, heart, and lung—also showed the Congo Red positivity diagnostic of AL, together with the discrete light-chain deposits along the basement membranes consistent with LCDD. The authors concluded that the same light chain was capable of forming either the fibrillar deposits of AL or the non-fibrillar deposits of LCDD, depending on environmental factors. There may, however, be no Congo Red positivity on renal biopsy, allowing the coexistence of AL to go unnoticed in the absence of other tissue samples [20].
In patients with multiple myeloma, MIDD may also occur in the presence of MCN. In a case series of 34 patients by Lin et al. on patients with MIDD, the biopsies with both MCN and LCDD were characterized by the significant tubular damage from the cast nephropathy, with less severe glomerular disease and less significant deposition of light chains along the basement membrane [8]. The clinical features and outcomes of these patients were more consistent with MCN patients than with pure MIDD patients. In another case series of 23 patients with LCDD and MCN, the glomeruli appeared normal in two-thirds of cases and nodular glomerulosclerosis was present in only three biopsies [11]. Again, the pathology was most notable for the tubular damage done by light chain casts, and the diagnosis of LCDD was made only by immunofluorescence. Of note, approximately one-third of cases did not demonstrate electron-dense deposits on electron microscopy. Lastly, in a recent series of 190 patients at the Mayo Clinic with multiple myeloma, 41 patients had MIDD and five had concurrent MCN and two had concurrent AL amyloidosis [21].
Lorenz et al. [22], in a recent case report, describe a patient who initially presented with hypertension and renal insufficiency and was found to have a monoclonal IgG kappa on serum immunofixation, and renal biopsy showed acute tubular necrosis, MCN, and focal arterial amyloidosis. She was treated with dexamethasone, thalidomide, and autologous stem-cell transplant and then had a recurrence 6 months later. Repeat biopsy revealed MCN, as well as Congo Red positive, positively birefringent deposits in the interstitium. Immunofluorescence demonstrated kappa light-chain positivity of the tubular basement membranes. Interestingly, no glomerular deposits were seen, and all three features—MCN, amyloidosis, and LCDD—were predominantly in the interstitium.
MIDD, though most often diagnosed by renal biopsy, is truly a systemic disease, with the possibility of immunoglobulin deposition in many tissues, including the heart, lung, and liver. Myocardial tissue demonstrates smooth, diffuse, and uniform staining in a perimuscular pattern [6, 10]. In lung tissue, there can be a more patchy deposition of eosinophilic material in alveolar walls, small airways, and vessels, with marked giant cell reaction. In MIDD, immunofluorescence shows either light or heavy chains within the basement membranes of the aforementioned structures, and electron microscopy demonstrates coarse extracellular granular deposits [23, 24]. Liver biopsy shows diffuse staining of the basement membranes, outlining the liver cell cords [6].
With treatment, there may be regression of the underlying nodular glomerulosclerosis and disappearance of the deposits [25, 26]. A case report describes a 53-year-old woman with LCDD who presented with nephrotic syndrome and nodular glomerulosclerosis on biopsy, received chemotherapy and an autologous stem-cell transplant, and achieved remission of her underlying disease. She then developed nephrotic syndrome 6 years later with a new IgG-lambda monoclonal gammopathy [27]. On repeat biopsy, she had IgG AH disease and no evidence of the prior nodular glomerulosclerosis. This case demonstrates the emergence of a second clone after autologous stem-cell transplant but also shows the reversibility of the glomerular lesion with effective treatment of the underlying disorder. Other authors, however, have reported persistence of light-chain deposits after chemotherapy and remission of underlying disease [28, 29].
Pathogenesis
The pathogenesis of MIDD is related to the primary structure of the protein itself, the amount of protein produced by a specific clone, the environment in which the immunoglobulin is presented, and the reaction of the cells in that environment. One thing that is clear from several large case series is that patients with multiple myeloma, and therefore those with the largest malignant clones, have a worse outcome than patients who simply have MGUS or no underlying plasma cell dyscrasia [2, 4, 6]. Though it is difficult to discern the exact cause for this discrepancy, one possibility is the amount of protein produced and presented to target organs dictates the severity of the disease. There are patients, however, with very aggressive and rapidly progressive MIDD in the absence of multiple myeloma, suggesting that it is more than just the absolute amount of immunoglobulin that dictates the severity of disease [30]. As LCDD is the most common subtype of MIDD, more is known about the pathogenesis of light chain, as opposed to heavy chain, deposition.
LCDD
In LCDD, the primary structure of the light chain likely has some importance in its ability to deposit in the body. Several proteins from patients with LCDD have been sequenced, and some conclusions can be drawn from analysis of the primary structure. The kappa light chains are more likely to be of the V-region subtype, of which Vκ[kappa]IV seems to be overrepresented, though κ[kappa]I–IV has been described [31–33]. There is no consistent structural motif or specific residue that has been shown to be responsible for the pathogenicity of these proteins; however, certain features have emerged. First, the amino acid substitutions appear to derive from somatic mutations, as opposed to germline mutations. Second, though the substitutions can occur throughout the protein, they are most common in the complementarity-containing region [31]. Third, the mutations described in both kappa and light chains are more likely to introduce hydrophobic residues, suggesting a disturbance in protein–protein interaction, destabilization of the protein, and subsequent deposition in tissues [31, 34, 35]. The predisposition to aggregation can be demonstrated in a murine model of LCDD, in which transfected mice with vectors containing kappa light chain sequence from a human subject with LCDD of the Vκ[kappa]IV subtype showed light-chain deposition in the kidney [36]. Lastly, posttranslational modification has also been implicated in creating pathologic light chains, as the isolate of one kappa light chain from a patient with LCDD had a mutation that resulted a new N-glycosylation site [37].
The mesangial cell is also an important factor in the pathogenesis of this disease. In a series of in vitro studies by Herrera et al., it has been demonstrated that mesangial cells are integral in the deposition of excess extracellular matrix (ECM) proteins that characterize the pathologic lesion of nodular glomerulosclerosis [38–40]. In one of the initial studies, cultured mesangial cells were incubated with light chains from patients with either amyloidosis, MCN, or LCDD [38]. The authors were able to show that mesangial cells cultured with amyloid light chains initially had an increase in ECM proteins, then a reduction to below control levels with a concurrent increase in collagenase activity. In contrast, mesangial cells cultured with LCDD light chains showed proliferation of mesangial cells followed by excess ECM formation, a reduction in collagenase activity, and mesangial nodules consistent with nodular glomerulosclerosis.
One important ECM protein that was present in these nodules was tenascin. Tenascin is a component of the mesangial ECM that is present in low levels in normal mesangium, but is present in much higher amounts in pathologic conditions of the kidney [41]. In addition, transforming growth factor β[beta] (TGF-β[beta]) is a known inducer of tenascin production and has been shown by the same authors to be present in the glomeruli of patients with LCDD [17]. Tenascin is degraded by the matrix metalloproteinase (MMP) 7. In the same in vitro model described above, the culture media from mesangial cells incubated with LCDD light chains showed a marked reduction in MMP-7 levels in comparison to those incubated with amyloid light chains [42] as well as an increase in TGF-β[beta] and platelet-derived growth factor β[beta] [40]. Interestingly, later studies have shown that MMP-7 is produced by mesangial cells, but the cells are for some reason unable to secrete it [40]. These experiments suggest that there is some intrinsic feature of the light chain itself that promotes or inhibits ECM production, alters regulation of ECM breakdown by MMPs and collagenase, and thereby produces different pathologic entities. One lingering question is how the same light chain is able to produce different types of deposits in the same individual or even in the same organ as described above (see Pathology).
HCDD
HCDD was first described as a distinct entity in 1992 by Tubbs et al. [43], where the proposed name was pseudo-γ[gamma] heavy-chain deposition disease. As mentioned above, the heavy chain in HCDD is more likely to be of the γ[gamma] class [8, 9], with some rarer cases of the α[alpha] subtype [16] and only one case of the μ[mu] subtype [44]. In most reported cases of γ[gamma]-HCDD, there is a deletion of the first constant (CH1) domain [18, 45–47], resulting in a truncated protein. One theory as to the mechanism of disease is that as these proteins lack the CHI portion, they cannot bind to the heavy-chain-binding protein (BiP) in the endoplasmic reticulum [48] and therefore are secreted into circulation rather than being degraded [49]. There may also be some amino acid substitutions in the complementarity-determining regions (CDRs) and framework regions that can change the property of the heavy chain, making it more likely to deposit in tissues [18, 50].
Further insight into the pathogenesis of HCDD can be gained by further subtyping the heavy chain. There have been reports of γ[gamma]1–4 subtypes [18, 43, 45, 49]. All of the γ[gamma]3 subtypes and the majority of the γ[gamma]1 subtypes reported have had low serum complements and deposition of C1q and C3 on immunofluorescence [49]. However, those with γ[gamma]4-HCDD and the one case of γ[gamma]2-HCDD did not have low serum complement or deposition of C3 and C1q. This is consistent with what is known about the classical pathway of the complement system, where IgG3 binds C1q most avidly, followed by IgG1 and IgG2 [51]. IgG4 fixes complement very poorly and is unable to activate the classical pathway. In a review of all the cases reported of γ[gamma]-HCDD, 11/19 patients had hypocomplementemia, as opposed to the five reported cases of α[alpha]-HCDD, where no patients had hypocomplementemia [16]. Interestingly, in all cases of α[alpha]-HCDD, crescents were observed on light microscopy. As crescents are rare in MIDD, this feature highlights the subtle but real pathogenic differences between all the subtypes of MIDD based on the characteristics of the deposited immunoglobulin. More research into the properties of the various immunoglobulins in MIDD may help explain the varied pathologic and clinical features of these different subtypes.
Clinical Presentation
The clinical presentation of MIDD depends on the organ systems involved and therefore can have a wide variety of signs and symptoms (Table 20.1). In general, affected individuals are in the sixth decade of life, and men are affected more often than woman in a ratio of 2:1 [2, 8, 13]. Given the varying criteria used for the diagnosis of multiple myeloma versus MGUS in several studies, the incidence of multiple myeloma in patients with MIDD is somewhat unclear. In the case of LCDD, the percentage is likely approximately 25–40 % [5, 6, 8], but has been reported as high as 58–65 % [2, 7, 9]. Conversely, patients with multiple myeloma have a 5–22 % lifetime incidence of LCDD. The incidence of MM in patients with HCDD and LHCDD may be slightly lower, 29 % and 50 %, respectively, in a recent case series [9]. The remainder of affected individuals may have MGUS with fewer than 10 % plasma cells on bone marrow biopsy, rarely another lymphoproliferative disorder, or no discernible hematologic disorder by conventional criteria.
Table 20.1
Summary of clinical characteristics of light-chain deposition disease
Kidney | Renal insufficiency |
Nephrotic syndrome | |
Proteinuria | |
Microscopic hematuria | |
Hypertension | |
Heart | Restrictive cardiomyopathy |
Congestive heart failure | |
Cardiac arrhythmias | |
Lung | Interstitial lung disease |
Pulmonary nodules | |
Cystic lung disease | |
Liver | Hepatomegaly |
Transaminase elevation | |
Fulminant hepatic failure | |
Nervous system | Peripheral neuropathy |
Focal brain lesions | |
Seizures | |
Retinal vasculopathy |
Serum protein electrophoresis (SPEP) is only positive in approximately 25 % of cases, but the majority of patients have an identifiable monoclonal protein by immunofixation in either the serum or urine, at a rate of 73–78 % and 79–90 %, respectively [2, 7, 13]. In one study of MIDD, the SPEP and urine protein electrophoresis (UPEP) were more likely to be positive in HCDD and LHCDD than LCDD, but in the patients with HCDD, the whole monoclonal Ig was detected by SPEP in only 4/7 patients and by UPEP in 1/7 [9]. The authors speculate that this may be due to its presence in very low titers or due to a rapid rate of deposition [52]. It is important to note that approximately 10 % of patients have no monoclonal protein detected by immunofixation, again emphasizing the importance of routine immunofluorescence on renal biopsy specimens. The diagnosis of MIDD by renal biopsy may precede the diagnosis of monoclonal gammopathy in up to 68 % of cases [8]. The newer free light-chain assay that quantifies the levels of light chains in the serum and gives a ratio of kappa:lambda has not only aided in the diagnosis of AL, LCDD, and other plasma cell dyscrasias but also correlates with disease activity and can be followed during the course of treatment [53, 54]. Table 20.2 demonstrates the clinicopathologic correlates of LCDD, LHCDD, and HCDD, respectively.




LCDD | HCDD | LHCDD | |
|---|---|---|---|
Light microscopy | |||
Nodular glomerulosclerosis | +++/+ | ++++ | +++/+ |
Global glomerulosclerosis | + | + | + |
TBM thickening | V | V | V |
GBM thickening | NS | NS | NS |
Crescents | + | + | + |
Tubular atrophy/interstitial fibrosis | V | V | V |
Immunofluorescence | |||
Anti-light chain on TBM | ++++ | ++++ | |
Anti-heavy chain on TBM | ++++ | ++++ | |
Anti-light chain on GBM | ++++ | ++++ | |
Anti-heavy chain on GBM | ++++ | ++++ | |
Electron microscopy | |||
Granular deposits along TBM | ++++ | ++++ | ++++ |
Granular deposits along GBM | ++++ | ++++ | ++++ |
Mesangial deposits | +++ | ++++ | ++++ |
Clinical features | |||
Creatinine at diagnosis (mg/dl) | 3.8–4.0 | 4.8–5.6 | 3.1–5.3 |
Nephrotic-range proteinuria (>3 g/day) | ++ | +++/+ | +/+ |
Nephrotic syndromea | + | +++ | +/+ |
Hypertension | ++++ | ++++ | +++/+ |
Diagnosis of multiple myelomab | +++ | ++/+ | +/+ |
+SPEP and immunofixation | ++/+ | +++/+ | ++++ |
+UPEP and immunofixation | +++/+ | +++/+ | ++++ |
+ = 1–25 %, ++ = 25–50 %, +++ = 50–75 %, ++++ = >75 %, values separated by / have range that span two quartiles
NS not significant, V variable with wide range
Related posts:





Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
