Mark L. Gonzalgo, MD, PhD, Alan Keith Meeker, PhD, MA In the United States alone, cancer strikes over half a million victims annually and is currently the second leading cause of death, poised to become the leading cause of death in the near future, should recent trends continue. Genitourinary (GU) malignancies comprise 28% (413,700) of all cancer cases in the United States (nonmelanoma skin cancers excluded) and 15% of all cancer deaths (Jemal et al, 2009). However, significant advances in the diagnosis and treatment of certain GU cancers have been made. For example, the cure rate for testicular cancer now approaches 100% (Einhorn, 2002; Horwich et al, 2006). Unfortunately, this cancer is unusual in its responsiveness to therapy and is relatively uncommon. We have had less success with the more prevalent GU malignancies, such as prostate, bladder, and renal cancers—the second, eighth and tenth most common cancers, respectively. Encouragingly, however, mortality figures for these malignancies have shown a slow but steady decline over the past decade, and there is every reason to believe that these trends will continue and even accelerate in the future. Much of our current understanding of cancer is the direct result of the molecular biology (Table 18-1) revolution that developed rapidly following the elucidation of the molecular structure of deoxyribonucleic acid (DNA) by Watson and Crick in 1953. In subsequent years, the field of molecular genetics has complemented and greatly expanded upon knowledge gleaned by other disciplines, such as biochemistry and cell biology, providing important insights, at the molecular level, regarding the abnormalities present in cancer cells. Thus a great deal is now known concerning the numerous molecular signaling pathways that provide both positive and negative regulatory signals that stringently control cell proliferation in normal cells, such that any losses in cell number are precisely counterbalanced by cell proliferation, thereby maintaining tissue and organ homeostasis. Renegade populations of autonomously proliferating cells represent a serious threat to survival of the organism, particularly to large, long-lived species such as ourselves; therefore, we have evolved multiple barriers to prevent such outbreaks from occurring. This means that incipient cancer cells must overcome several hurdles on the way to becoming fully malignant—a multistep process that takes many years or even decades to complete. Consequently, it has been recognized that cancer cells need to acquire several key attributes in order to make the transition from a normal cell to a malignant tumor. These attributes include (1) genetic instability, (2) autonomous growth, (3) insensitivity to internal and external antiproliferative signals, (4) resistance to apoptosis and other forms of induced cell suicide, (5) unlimited cell division potential, (6) the ability to induce new blood vessel formation, a process termed angiogenesis, (7) locally invasive behavior, which uniquely distinguishes malignant from benign neoplasms, and (8) evasion of the immune system. In addition, cancer cells need to cope with various cellular stresses that are byproducts of their abnormal physiology. Finally, many cancers develop an additional, lethal attribute—the ability to leave the site of the primary tumor to colonize and thrive in distant organs or tissues as metastases (Hanahan and Weinberg, 2000; Solimini et al, 2007; Luo et al, 2009). Table 18–1 Molecular Biology: Glossary of Terms
| Allele: An alternative form of a gene. |
| Amplification: Additional copies of a chromosomal sequence; these sequences may include genes and may be extrachromosomal or intrachromosomal. |
| Aneuploid: Deviation in chromosomal number from the usual diploid state (e.g., tetraploidy) |
| Annealing: The pairing of two single strands of complementary DNA sequences to form a double helix. |
| Alternative splicing: A mechanism by which variations in the incorporation of a gene’s coding regions during mRNA maturation leads to the production of multiple related forms of a gene. |
| Base pair: The physical relationship between adenine/thymidine and guanine/cytosine within the double helix of DNA. Abbreviated as bp, it provides the unit of measurement for DNA. Each base pair is stabilized by hydrogen bonds. |
| cDNA: A segment of DNA complementary to an RNA sequence. |
| Centromere: An essential structural element of chromosomes consisting of repetitive, non-coding DNA to which the spindle microtubules attach during mitosis. |
| Chromosome: A distinct segment of linear DNA containing a large number of genes. In humans there are 23 such segments, each containing hundreds to thousands of genes. |
| Codon: Three sequential nucleotides in protein coding genes that specify a particular amino acid in the protein or a STOP translation signal. |
| Deletion: The removal of a segment of DNA, with rejoining of the ends. |
| Diploid: A set of chromosomes containing two complete copies of the organism’s genomic DNA. |
| Dominant: Allele determining the phenotypic manifestation of a gene. |
| Endonuclease: An enzyme that cuts DNA or RNA within the nucleotide chain. |
| Epigenetic: Nongenetic information, such as methylation or acetylation of histone proteins that modifies gene expression. |
| Exon: A sequence of DNA that is represented with a complementary RNA sequence, also known as the coding sequence. |
| Expression vector: A vector designed to encode a particular DNA sequence or gene for transcription and translation into RNA or protein. |
| FISH: Fluorescence in situ hybridization. A technique in which a fluorescently-labeled nucleic acid probe is hybridized to its complementary target sequence in the genome, allowing localization and enumeration of the target. |
| Frameshift mutation: A deletion or insertion of DNA that shifts the normal codons into a different order for translation into protein. |
| Gene: A segment of DNA that contributes to the formation of a protein, including both the introns (noncoding regions) and the exons (coding regions), as well as the regulatory regions preceding and following the coding regions. |
| Genetic code: The correspondence between triplets of DNA or RNA (codons) and amino acids making up proteins. |
| Genotype: The genetic makeup of an organism as reflected in its DNA sequence. |
| Haplotype: A group of alleles in relative close proximity on a chromosome that are inherited together. |
| Hemizygote: Having only one copy of a gene owing, for example, to the loss of chromosomal material or an entire chromosome. |
| Heterozygote: Having two different copies (alleles) of a gene. |
| Homozygote: Having two identical copies (alleles) of a gene. |
| Hybridization: The physical pairing of complementary RNA and/or DNA sequences. |
| Intron: A segment of DNA that is transcribed but is removed by the splicing together of the exons on either side; part of the noncoding sequences of a gene. |
| Karyotype: A catalogue of all the chromosomes within a particular cell. Typically accomplished by isolation and staining of metaphase chromosomes. |
| Linkage: The tendency for genes in proximity to one another on a chromosome to be inherited together. |
| Locus: The position of a particular gene on a particular chromosome. |
| Loss of heterozygosity (LOH): Deletion or mutation of a gene creating a hemizygous state or, if the remaining allele is duplicated, a homozygous state. In cancers arising in cancer predisposition syndromes caused by an inherited gene mutation, the remaining wild-type allele is often lost by LOH. |
| Methylation: The addition of a methyl group to a molecule. Methylation of the nucleotide cytosine in the promoter regulatory region of a gene is often associated with decreased transcription of that gene. |
| Micro-RNA: (miRNA); small, single-stranded RNA molecules, 21 to 23 nucleotides in length, that function to negatively regulate transcribed genes for which they have sequence complementarity. |
| Mutation: Any change in the sequence of genomic DNA. |
| Northern blotting: A technique of transferring RNA from an agarose gel to a nitrocellulose filter for hybridization with a complementary DNA. |
| Oncogenes: Genes that encode for proteins that have the ability to transform normal cells into cancerous cells. |
| Phenotype: The appearance or function of an organism, reflecting the contributions of the genotype and the environment. |
| Ploidy: The number or copies of entire chromosome complements (genomes) within a cell; diploid has two copies, triploid has three copies, tetraploid has four copies, etc. |
| Point mutation: A change in a DNA sequence involving a single base pair. |
| Polymerase chain reaction (PCR): A technique using sequential temperature cycles favoring DNA denaturation, annealing of primer sequences, and primer extension with DNA polymerase to amplify a large number of copies of a particular sequence of DNA. This technique can be used to detect very small amounts of DNA by creating a huge number of identical copies. |
| Polymorphism: A difference in normal DNA sequence between individuals. It can be a repetitive element or a single nucleotide polymorphism (SNP). |
| Promoter: The region of DNA in a gene where RNA polymerase binds and gene transcription begins. This region is often the sequence of DNA 100 to 500 base pairs immediately before the initiation site of the protein coding portion of the gene. |
| Recessive: An allele that is not represented phenotypically in the presence of a dominant allele. |
| Reporter gene: A gene encoding for a new or foreign protein that can easily be detected. For example, the luciferase gene, encoding the light-producing proteins of the firefly, is introduced into cells that do not express this gene. |
| Restriction enzyme: An enzyme recognizing specific short sequences of DNA (e.g., 4 or 6 base pairs); these enzymes cut the DNA at a particular location. |
| Silent mutation: An alteration in a DNA sequence that does not change the protein product of the gene. |
| Small interfering RNA: (siRNA); small double-stranded RNA molecules, 20 to 25 nucleotides in length, that promote degradation of mRNA molecules to which they have complementarity, thus downregulating expression of the corresponding gene—often employed by researchers as a means of reducing expression of specific genes. |
| Southern blotting: A technique of transferring denatured DNA from an agarose gel to a nitrocellulose filter for hybridization with complementary DNA. |
| Splicing: The removal of introns (spliced out) and the connection of exons (spliced together) in mRNA. |
| Telomere: An essential structural element of chromosomes consisting of repetitive, noncoding DNA and associated telomere-specific binding proteins that cap and stabilize the chromosomal termini. |
| Transcription: Synthesis of RNA on a DNA template. |
| Transfection: The introduction of DNA sequences into a cell. |
| Transgenic animals: Animals created by the introduction of new DNA into the germ line (into the egg). |
| Translation: Synthesis of protein from the messenger RNA (mRNA) template. |
| Vector: A plasmid, bacteriophage, or virus that carries new DNA into a cell. Vectors are often designed to produce large amounts of protein encoded by the gene within the host cell. |
Basic Molecular Genetics
DNA
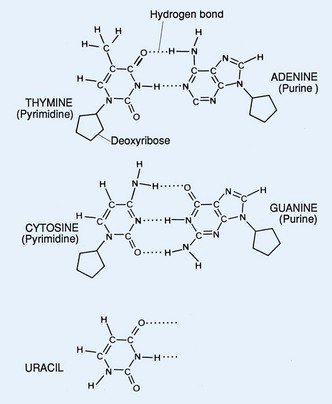
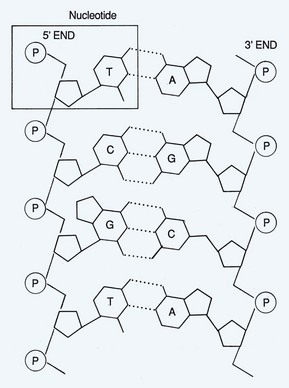
The molecular characteristics of DNA were first described in 1953 (Watson and Crick, 1953). This molecule serves as the blueprint for determination of structure and function of all living organisms. DNA is composed of three basic components: a pyrimidine or purine base, a sugar (2-deoxyribose), and a phosphate (Fig. 18–1). The connection between repeating phosphates and sugars creates a helical chain. The DNA structure exists as a double helix in which one strand of bases is ordered in one direction and the other strand is ordered in the opposite direction. The two strands are held together by hydrogen bonds and are organized by complementary base pairing. The four bases that primarily comprise DNA are adenine, cytosine, guanine, and thymine. There is a fifth base that can be found in DNA known as 5-methylcytosine (5-mC). Uracil is substituted for thymine in ribonucleic acid (RNA). Hydrogen bonding occurs specifically between the purine adenine (A) and the pyrimidine thymine (T) and between the purine guanine (G) and the pyrimidine cytosine (C) (Fig. 18–2). In the RNA molecule, adenine base pairs with uracil (U).
Transcription
During the process of transcription, linear DNA is converted to linear messenger RNA (mRNA). The process of translation consists of the conversion of linear mRNA to a linear set of amino acids that will eventually form a functional protein (Fig. 18–3). Transcription is the first step in converting DNA into protein. A single strand of RNA is copied from one of the strands of DNA. The sugar element in the RNA molecule is ribose and the pyrimidine uracil substitutes for thymine. RNA polymerase II is the enzyme that synthesizes the first copy of RNA, which is a complementary strand of the DNA template. This primary strand of RNA is called heterogeneous nuclear RNA (hnRNA) and contains coding sequences (exons) of DNA and noncoding sequences (introns).
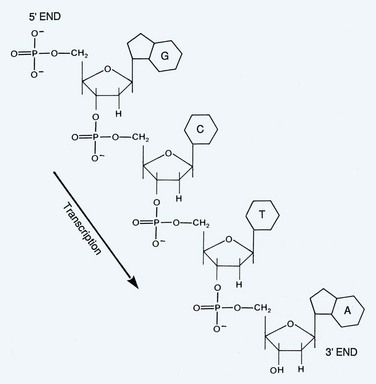
The pre-mRNA molecule undergoes three major modifications: 5′ capping, 3′ polyadenylation, and RNA splicing. These processes occur in the nucleus before the RNA molecule is translated into protein. A guanine is added to the 5′ end of the molecule which results in the formation of a methylated cap (Perry, 1981). A long chain of approximately 200 adenine residues forms what is called a poly(A) tail that is added to the 3′ end of the mRNA and may provide stability to the molecule (Jackson and Standart, 1990). RNA splicing is a dynamic process that can result in the formation of different peptide sequences. A primary RNA transcript is converted into a mature mRNA prior to protein synthesis. The process known as alternative RNA splicing is how a single gene can encode for multiple unique proteins by including or excluding certain exons in the mRNA transcript (Sharp, 1987). The sequences contained within introns are not essential for protein synthesis; therefore introns are spliced out and exons are spliced together to generate the functional message known as mRNA. Exons are sequences of DNA that have corresponding sequences in the mRNA, and introns are regions of DNA sequence that are not represented in mRNA (Witkowski, 1988).
RNA Interference
Post-transcriptional gene regulation can occur by a mechanism involving the expression of noncoding RNAs that have the capability of binding to and degrading messenger RNAs. RNA interference (RNAi) was first discovered in Caenorhabditis elegans, but exists as a means of post-transcriptional regulation in a wide variety of organisms (Fire et al, 1998). RNAi are double-stranded RNAs that are transcribed from cellular genes or infecting pathogens. A double-stranded RNA species is reduced in size by a ribonuclease (RNAse) III–like enzyme, known as Dicer, into 21– to 23–base pair (bp) products with two-nucleotide 3′ overhangs known as small (or short) interfering RNA (siRNA) (Bernstein et al, 2001). These short RNA molecules are homologous to the gene that is being suppressed. The antisense strand of the siRNA is incorporated into a multicomponent nuclease called “RNA-induced silencing complex” (RISC) to destroy specific mRNAs (Gregory et al, 2005).
RNAi has been used to investigate gene function in the laboratory. Long micro-RNAs contain self-complementary regions resulting in the formation of hairpin loops that are cleaved by two RNAse III–like enzymes. The enzyme called Drosha cuts the long hairpin to generate a short hairpin RNA (shRNA) molecule. The shRNA is then processed by Dicer to produce double-stranded miRNA capable of binding target genes for destruction. Synthetic siRNA and shRNA species can be generated to target and knockdown specific gene expression. For example, this technique has been used to study the role of the androgen receptor (AR) in the progression of castrate-resistant prostate cancer. Expression of AR was knocked down using an shRNA in prostate cancer xenografts and cell lines, demonstrating that AR was necessary and sufficient for tumor growth, thereby implicating continued AR function as an essential component in both hormone-dependent and castrate-resistant prostate cancer (Chen et al, 2004).
Protein Translation
Translation of mRNA into protein occurs in the cytoplasm where ribosomes are located. The mRNA message is translated in segments of three adjacent nucleotides called a codon. Each codon is translated into one of 20 amino acids (Fig. 18–4). A total of 64 different codons can be generated from the four bases found in RNA. Due to redundancy, more than one codon can encode for a single amino acid (Fig. 18–5). Genetic mutations or alterations can have a profound effect on protein translation of mRNA. For example, a single base insertion or deletion results in a frame-shift mutation and often results in a nonfunctioning protein product. In contrast, a single base substitution may or may not result in a change in the amino acid sequence to alter protein function.
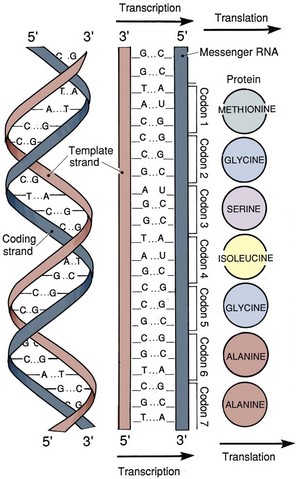
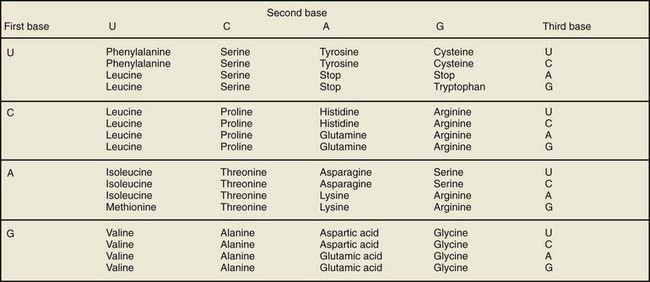
The three major stages of protein synthesis are: initiation, elongation, and termination. Initiation is frequently the rate-limiting step in translation (Merrick, 1992). During the process of initiation, the 40S ribosome binds to mRNA and forms an initiation complex that binds a tRNA with an amino acid attached. All new proteins start with methionine because the initiation codon is AUG. During elongation, amino acids are sequentially added to the peptide chain, and the ribosome moves along the mRNA chain similar to beads on a string. Elongation is the most rapid phase of protein synthesis. For any given mRNA transcript, multiple ribosomes can simultaneously move along the chain to generate new polypeptides (Rich et al, 1963). Termination is the last stage of protein synthesis and is signaled by one of three “stop” codons (Tate and Brown, 1992). The release factor is a cytoplasmic protein that binds to the stop codon and catalyzes the hydrolysis of the aminoacyl linkage that releases the carboxyl end of the polypeptide chain (Merrick, 1992). The ribosome releases from the mRNA and dissociates into two subunits after termination.
Tumor Suppressor Genes and Oncogenes
Tumor Suppressor Genes
Tumor suppressor genes regulate cellular growth and play a critical role in the normal processes of the cell cycle. These genes are also important for DNA repair and cell signaling. The absence of tumor suppressor gene function may lead to dysregulation of normal growth control and malignancy. Loss of function of both alleles of a tumor suppressor gene is typically required for carcinogenesis. This functional loss can occur by (1) homozygous deletion, (2) loss of one allele and mutational inactivation of the second allele, (3) mutational events involving both alleles, and (4) loss of one allele and inactivation of the second allele by DNA methylation. Classic tumor suppressor genes discussed later in this chapter are the retinoblastoma gene (RB) and TP53 (see The Cell Cycle).
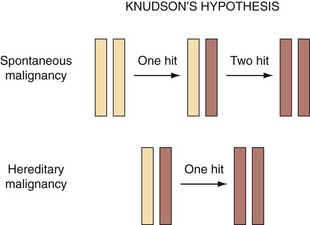
The “two-hit” hypothesis was first proposed in cases of retinoblastoma, which required mutations in both alleles for disease manifestation (Knudson, 1971). This is because if just one allele is inactivated, the remaining allele could produce a sufficient amount of the correct protein to maintain the normal state (Fig. 18–6). Specific types of mutations in certain genes, however, may not follow this two-hit rule and can function as dominant negative mutations that produce altered protein. Mutant protein products have been reported to inhibit function of normal protein from unaltered alleles (Baker et al, 1990). Mutation of a single allele may result in haploinsufficiency, causing increased carcinogen susceptibility as in the case of the TP27Kip1 gene (Fero et al, 1998). Abnormally low levels of the TP27 protein were frequently observed in human cancers; however, homozygous inactivating mutations of the TP27 gene were infrequent. Both Tp27 nullizygous and Tp27 heterozygous mice were predisposed to developing tumors when exposed to radiation or a chemical carcinogen. The assumption that null mutations in tumor suppressor genes are recessive excludes genes that exhibit haploinsufficiency (Fero et al, 1998).
Oncogenes
Oncogenes are associated with cellular proliferation and are the mutated form of normal genes (proto-oncogene). A number of oncogenes were initially identified by their presence in retroviruses that were capable of inducing malignant transformation of normal cells (Martin, 1970). Two oncogenes that have been found to be overexpressed in a variety of cancers include c-MYC and c-MET (Wong et al, 1986; Bottaro et al, 1991). The proto-oncogene, c-MYC, encodes an early-response gene product that is a transcription factor responsible for regulating cellular proliferation. Amplification of c-MYC is a frequent event in prostate cancer and expression of c-MYC in human prostate epithelial cells has been associated with immortalization (Gil et al, 2005).
Hepatocyte growth factor acts through a receptor that is encoded by the proto-oncogene c-MET (Bottaro et al, 1991). Hepatocyte growth factor plays a role in normal development of the kidney, acts as a mitogen for renal tubular cells, and is capable of inducing tubulogenesis of renal cells in vitro (Montesano et al, 1991; Santos and Nigam, 1993). Increased expression of c-MET has been reported in renal cell carcinoma and is also more frequent in higher grade cancers (Pisters et al, 1997). Missense mutations of the MET proto-oncogene may also result in constitutive activation of the MET protein in tumors associated with hereditary renal cell carcinoma (Schmidt et al, 1997).
Mechanisms by which a proto-oncogene can be converted to an oncogene are by (1) mutation of the proto-oncogene resulting in an activated form of the gene, (2) gene amplification, and (3) chromosomal rearrangement. A mutation occurring within the coding sequence of a gene can lead to a continuous proliferation signal. The proto-oncogene ERBB encodes for the epidermal growth factor receptor (EGFR), and mutations of ERBB can result in overexpression of a receptor form that is constitutively active (Downward et al, 1984). Errors that occur during chromosomal replication may result in gene amplification and aneuploidy. An increase in gene copy number can produce an increased number of mRNA transcripts and overproduction of protein. For example, certain types of bladder cancer have been found to over-express c-MYC by this mechanism (Christoph et al, 1999). Immunohistochemical staining of bladder cancer specimens has demonstrated overexpression of c-MYC protein in more than half of papillary and invasive tumors (Schmitz-Drager et al, 1997). Finally, chromosomal rearrangements such as translocation events can result in the formation of an oncogene. This has been classically described in chronic myelogenous leukemia in which the Philadelphia chromosome is formed by reciprocal translocation between chromosome 9 and 22 (Nowell and Hungerford, 1960). A portion of the breakpoint cluster region (BCR) gene from chromosome 22 (region q11) is juxtaposed to the ABL1 gene on chromosome 9 (region q34) to form a constitutively active BCR-ABL transcript.
Key Points
Tumor Suppressor Genes and Oncogenes
The Cell Cycle
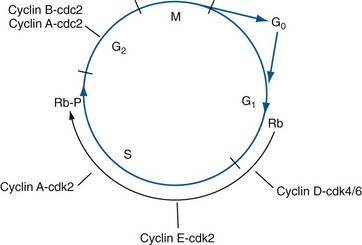
Quiescent cells are considered to be out of cycle, in a reversible state known as “G0,” which is the default state for most cells (Fig. 18–7). When signaled to proliferate, cells activate their cell cycle machinery, initiating an orderly, unidirectional, series of events resulting in (1) accurate duplication of the cell’s genome during the DNA synthetic phase (S phase), followed by (2) accurate segregation of each genomic complement to each of two resulting daughter cells, a process referred to as mitosis (M phase). These two critical phases are separated by two so-called “gap” phases (G1 and G2). Throughout the cell cycle, which takes approximately 24 hours to complete, each sequential step is dependent upon completion of the prior step before progressing further (Hartwell et al, 1974). In addition, checkpoint mechanisms closely monitor DNA integrity as well as certain critical cell cycle events. If problems are detected (e.g., DNA damage), the cell cycle will pause to allow repair (Hartwell and Weinert, 1989). If repair is not possible, normal cells often will commit cellular suicide through an active process termed apoptosis. In addition to proliferative signaling networks, many oncogenes and tumor suppressors exert their effects by interfering with cell cycle checkpoints and apoptotic pathways, allowing cancer cells to divide continuously and accumulate. Loss of the ability to respond appropriately to damaged DNA is particularly dangerous, because it fosters genetic instability, a key attribute of cancer cells. Loss of DNA damage checkpoint controls results in an increased mutation rate, accelerating the mutation of cancer-associated genes, thus contributing to carcinogenesis and disease progression (Bartek et al, 1999).
Cyclin-Dependent Kinases and Cyclins
The temporal sequence of events occurring throughout the cell cycle are effected by a highly conserved set of serine- and threonine-specific protein kinases termed cyclin-dependent kinases or CDKs for short (Meyerson et al, 1992). CDKs phosphorylate specific protein substrates involved in executing the phase-specific activities of the cell cycle. As their name implies, the enzymatic activities of the CDKs are dependent upon a class of regulatory proteins called cyclins, of which there are 12 known in mammals (Morgan, 1995). Cyclins, so named because the abundance of specific cyclins within the cell was found to be highly dynamic and tightly linked to specific phases of the cell cycle, physically associate with and activate CDK enzymatic activity (De Bondt et al, 1993; Jeffrey et al, 1995) (Fig. 18–7). Additionally, cyclins also assist in guiding CDK substrate specificity. Thus depending largely on the transitory presence of particular cyclins, which are rate-limiting, specific cyclin/CDK complexes form and act during restricted periods within the cell cycle to initiate and regulate the events required during these precise phases, after which each cyclin is typically degraded by polyubiquitination of specific lysine residues that serve as the signal for proteosomal degradation, thereby inactivating its CDK partner (Sherr, 1993; van den Heuvel and Harlow, 1993). The rapid, phase-specific destruction of cyclins also underlies the irreversible nature of the cell cycle. CDKs are also regulated by specific sites of phosphorylation on the CDKs themselves (Lundgren et al, 1991). For example, activation of the cyclin-dependent kinase CDK2 is dependent upon phosphorylation, by CDK-activating kinase (CAK), of threonine 160, which moves a regulatory loop away from the CDK2 catalytic active site, thereby allowing access to substrate. In contrast, phosphorylation of tyrosine 15 on CDK2 (effected by kinases WEE1 or MIK1) acts to inhibit CDK2 enzymatic activity. In addition, removal of this inhibitory phosphate group can occur by the phosphatase CDC25 (Sebastian et al, 1993). Finally, a group of proteins termed cyclin-dependent kinase inhibitors (CDKIs) exist that can bind to and directly inhibit CDK activity or their activating phosphorylations (Peter and Herskowitz, 1994; Sherr and Roberts, 1995). Thus, although cyclins play major regulatory roles in orchestrating CDK activities, CDKs are subject to additional levels of control and, as might be expected, these processes are also commonly altered in cancer cells.
Cell Cycle Entry
Under normal conditions a cell will be induced to proliferate when the balance between growth-stimulatory signals received from its environment (e.g., soluble growth factors in the extracellular space) outweighs growth inhibitory signals (e.g., TGF-β). If the cell was in the G0 state, it will proceed into G1, the first gap phase of the active cell cycle, during which sufficient macromolecules and organelles are synthesized to support cell replication. Growth stimulatory signaling pathways (e.g., the RAS/RAF/MEK pathway) promote expression of the main G1 cyclins, cyclin D and cyclin E, leading to their association with CDK4 and CDK6 kinases, thus activating these CDKs (Baldin et al, 1993; Winston and Pledger, 1993; Sherr et al, 1994). Throughout early and mid-G1, progress through the cell cycle remains sensitive to external signals, largely because persistent mitogenic stimulation is required for sustained expression of cyclin D (Matsushime et al, 1991). Cyclin D is the main cyclin functioning early in G1. Cyclin E is expressed later in G1, peaking at the G1S boundary, where its presence is required for S-phase entry (Dulic et al, 1992; Koff et al, 1992; Ohtsubo et al, 1995).
The Retinoblastoma Protein and the Restriction Point
Due to the dependence of cyclin D on the continued presence of mitogens and other favorable conditions (e.g., attachment to the substratum), the cell cycle remains responsive to external signals throughout early and mid-G1 (Guadagno et al, 1993). However, if the decision is made in late G1 to continue on with cell division, then the subsequent phases of the cell cycle (S, G2, and M) proceed in an autonomous fashion. This key point in time in late G1, after which the cell cycle becomes insensitive to extracellular signals, is termed the restriction point (R point) and is highly regulated (Pardee, 1974).
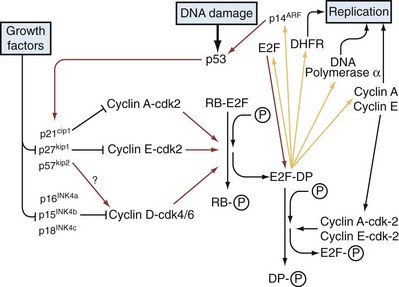
The retinoblastoma susceptibility protein, pRB, plays a central role in controlling the R point, and the inappropriate, continuous proliferation of cancer cells is largely due to a loss of R-point control, typically the result of functional inactivation of the RB pathway (Pardee, 1989). pRB is a member of a protein family termed pocket proteins that includes the closely related proteins TP107 and TP130. pRB is a substrate of G1 cyclin/CDK complexes and its activity is critically dependent on its phosphorylation state (Buchkovich et al, 1989; Mittnacht et al, 1994; Sherr, 1994). In nondividing cells, pRB is unphosphorylated and in this state pRB and the other pocket proteins physically associate with members of the E2F family of transcription factors, inhibiting their transcriptional activation of target genes required for cell cycle progression into S phase (Lai et al, 1999b). In addition, pRB recruits proteins such as histone deacetylases (HDACs) to E2F target genes promoting the formation of repressive heterochromatin at these sites and pRB itself may play a role in maintaining this repressive chromatin state, further suppressing E2F target genes (Lai et al, 1999a, Gonzalo and Blasco, 2005). However, during G1, pRB becomes progressively phosphorylated, on 8 different sites, by cyclin D-CDK4/6 and cyclin E-CDK2 complexes, creating hyperphosphorylated pRB, causing its dissociation from E2F (Fig. 18–8). The liberated E2F proteins, of which there are at least seven, heterodimerize with one of three DNA polymerase (DP) subunits (DP1-3) and activate transcription of target genes, including essential S-phase genes (e.g., DNA polymerase and S-phase cyclins) (Dyson, 1998). Notably, the cyclin E gene is an E2F target gene; thus once E2Fs become activated, a positive feedback loop is initiated, with further production of cyclin E reinforcing pRB hyperphosphorylation, which, in turn, frees additional E2F. In addition, activated E2F increases transcription of its own gene, thus providing another positive feedback loop. These autocatalytic cycles represent the molecular core of the R point. Beyond this point, pRB remains inactivated and the remaining cell cycle phases are unresponsive to either external mitogenic or antiproliferative signals. Once the cell cycle is complete, pRB returns to its unphosphorylated state, once more suppressing E2F until cell division is once again signaled (Ludlow et al, 1993).
The pRB gene was initially characterized as the tumor suppressor gene responsible for disease susceptibility in familial retinoblastoma (thus the name) and, as is the case with many cancer predisposition genes, was also found to be compromised in sporadic retinoblastoma as well (Friend et al, 1986; Knudson, 1993). As outlined above, its central role as the gatekeeper of the R point in the cell cycle explains why pRB is also commonly altered in other cancers besides retinoblastoma. In fact, it has been suggested that RB, or components of the regulatory pathway it is associated with (e.g., cyclins D and E, CDK4, E2F, TP16 and TP27), are likely to be altered in virtually all human cancers (Sherr, 1996; Hanahan and Weinberg, 2000).
Retinoblastoma Protein and Genitourinary Malignancies
pRB gene mutations have been identified in approximately one third of bladder tumors (Horowitz et al, 1990), and reintroduction of the pRB gene into bladder carcinoma cell lines has been found to inhibit cell growth in vitro and tumor formation in vivo (Takahashi et al, 1991). Altered expression of pRB protein has been also identified in approximately one third of bladder carcinomas (Logothetis et al, 1992), and altered expression has been correlated with higher-stage disease and decreased patient survival (Cordon-Cardo et al, 1992).
Prostate carcinoma has not been as strongly linked to pRB. Although pRB mutations are present in 10% to 30% of prostate cancer specimens (Bookstein et al, 1990; Kubota et al, 1995), decreased expression is not consistently identified with high-risk patients or recurrent disease (Kibel and Isaacs, 2000). In other studies, no correlation was found between expression and grade or stage (Ittmann and Wieczorek, 1996), but Theodorescu and colleagues (1997), reported that low pRB protein expression correlated with decreased disease-specific survival in univariate and multivariate analysis.
Renal carcinoma has not been clearly linked to pRB. pRB is rarely inactivated in renal carcinoma cell lines or tumors (Ishikawa et al, 1991), and analysis of clinical specimens has not demonstrated a clear association between prognosis and pRB expression (Lipponen et al, 1995).
Cell Cycle Progression through S phase
During S phase, a normal cell replicates its diploid genome once and only once. S-phase genes are activated as a result of cyclin A replacing cyclin E to form cyclin A-CDK2 complexes in early S phase, and later by cyclin A associating with CDK1 forming cyclin A-CDK1 complexes in late S phase (Girard et al, 1991; Pagano et al, 1992). Upon completion of S phase, cyclin A-CDK2 phosphorylates E2F-DP heterodimers, thus terminating E2F’s activity (Dynlacht et al, 1994). At the end of S phase, cyclin B replaces cyclin A, forming cyclin B-CDK1 complexes that move the cell into the second gap phase of the cell cycle, G2, and on into mitosis (Pines and Hunter, 1989). It should be noted that the above description, although correct in outline, has been simplified. For example, there are actually multiple closely related cyclins within each cyclin class; thus there are actually three D-type cyclins (D1, D2, and D3), two E-type cyclins (E1 and E2), two A-type cyclins (A1 and A2), and two B-type cyclins (B1 and B2).
Mitosis
Although mitosis is the shortest of the cell cycle phases (lasting approximately 1 hour), it is one of the most complex and dramatic (Alberts, 2007). During mitosis, the two genomic complements present following S phase are precisely partitioned into what will become two separate daughter cells. The proper functioning of many of the proteins participating in mitosis depends upon phosphorylation of these substrates by cyclin B-CDK1. Early in mitosis, the chromosomes condense through the activity of condensin proteins. Each chromosome is tightly associated along its length with its corresponding S-phase copy, termed the sister chromatid, through cohesin proteins. When isolated and stained, these condensed metaphase chromosomes are used in karyotyping studies to look for numerical and structural chromosome aberrations. During this time also, two structures called centrosomes, which act as microtubule organizers, migrate to opposite poles of the nucleus and assemble spindle microtubules that radiate through the nucleus and attach to the condensed chromosomes at constriction sites termed centromeres, through complex protein matrices termed kinetochores. The nuclear envelope and supporting intermediate filament proteins, the nuclear lamins, dissociate in response to phosphorylation by cyclin B-CDK1. Balanced tension along the spindle microtubules causes all of the chromosomes to align along the cell midline, creating a hallmark stage of mitosis termed metaphase. Once all metaphase chromosomes are properly aligned, a protein complex termed the anaphase promoting complex (APC, not to be confused with the adenomatous polyposis coli gene, which is commonly mutated in colon cancer) is activated by cyclin B-CDK1. The APC, in turn, ubiquitinates a protein called securin, targeting it for destruction, which liberates separase protease from securin’s inhibitory influence (Nasmyth et al, 2000). Once activated, separase functions to cleave the cohesin complexes, thus liberating the sister chromatids from one another, allowing microtubule-associated motors on the spindle microtubules to pull each diploid chromosome complement apart toward the centrosomes at opposite poles, a key mitotic event termed anaphase. One of the APC’s substrates is cyclin B itself; thus by activating the APC, cyclin B initiates its own destruction (Deshaies, 1995). Lastly, two new nuclei are assembled around the two separated chromosome complements, which decondense, and two separate daughter cells are formed in a cell-splitting process termed cytokinesis.
Mitotic abnormalities are commonly seen in human cancers and were noted by David Hanseman in the late 1800s, soon after the discovery of chromosomes, and, in 1914, the eminent German biologist Theodor Boveri proposed that such abnormalities might underlie tumor formation (Harris, 2008).
Cyclin-Dependent Kinase Inhibitors
In addition to their dependence on cyclins and regulation by specific phosphorylations, CDK activities are also subject to negative regulation by a set of seven proteins termed cyclin-dependent kinase inhibitors (CDKIs). CDKIs belong to either of two different classes: the CIP/KIP family, which includes the proteins TP21 (CDKN1A), TP27 (CDKN1B), and TP57 (CDKN1C); and the INK4 (inhibit CDK4) family, which includes TP15 (INK4B), TP16 (INK4A), TP18 (INK4C), and TP19 (INK4D). The CIP/KIP proteins are broadly acting, able to inhibit multiple cyclin-CDK complexes throughout the cell cycle (Clurman and Porter, 1998), while the members of the INK4 group are more restricted in their activities, inhibiting CDK4 and CDK6-containing complexes; thus they are critical regulators of the R point and the G1S transition because they can block pRB phosphorylation (Fig. 18–8). Increased expression and accumulation of CDKIs is used by the cell as a means of halting the cell cycle in response to various stresses. For example, TP21 expression is increased in response to DNA damage (el-Deiry et al, 1993). CDKIs function in nonstress situations as well. For example, TP27 levels are high in quiescent cells, and all of the CIP/KIP proteins appear to play some role in maintenance of the G0 state in terminally differentiated cells (Halevy et al, 1995; Matsuoka et al, 1995). Interestingly, mice engineered with a homozygous deletion of the TP27 gene grow more quickly, exhibit hyperplasia in multiple organs, and are cancer prone (Fero et al, 1996). In addition to increased expression in order to inhibit cell cycle progression, CDKIs can also be oppositely regulated by proteolytic degradation—for example, the targeted elimination of TP27 upon mitogenic stimulation (Ohtsubo et al, 1995).
Among the INK4 family, inactivating mutations and abnormal methylation of TP15 and TP16 have been strongly implicated in cancer in general (Kamb et al, 1994; Hirama and Koeffler, 1995) and specifically in genitourinary malignancies (Cairns et al, 1995; Herman et al, 1995). The best studied of the INK4 proteins is TP16. The gene was initially found mutated and deleted in a wide variety of tumors including bladder and kidney (Kamb et al, 1994). Subsequent analysis has demonstrated that inactivation often occurs by DNA hypermethylation at the TP16 promoter, an alternative method of gene inactivation (Merlo et al, 1995). Interestingly, the same CDKN2A locus codes for an entirely different gene product, TP14ARF, the coding sequence of which partially overlaps with the TP16 coding sequence but uses a separate gene promoter and is translated using an alternate reading frame (Quelle et al, 1995). TP14 modulates the TP53 tumor suppressor pathway, serving to stabilize TP53 protein by inhibiting the ubiquitin ligase HDM2 (Mdm2 in the mouse) which normally tags TP53 for destruction. TP14 binds to HDM2 directly inhibiting it, as well as sequestering it in the nucleolus, a subcompartment within the cell nucleus (Honda and Yasuda, 1999; Llanos et al, 2001). In addition, TP14 can also bind to E2F transcription factors, inhibiting them and inducing their degradation (Rizos et al, 2007). The TP15 gene lies just adjacent to the TP16 gene on the short arm of chromosome 9 (9p21), and therefore was deleted in many of the same tumors and cell lines that lost TP16 (Hannon and Beach, 1994). However, TP15 does not appear to play as strong a role in tumorigenesis as TP16 (Stone et al, 1995).
The TP16 gene is frequently inactivated by deletion in bladder carcinomas (Cairns et al, 1995; Williamson et al, 1995). Importantly, despite its proximity, the TP15 gene was ruled out as the primary tumor suppressor at this site because it was not within the deletion interval. A study by Orlow and colleagues (1999) found that deletion and methylation of the TP16 gene occurred frequently in superficial bladder carcinoma, but only those deletions that affect both the TP16 and TP14ARF genes correlated with a decrease in disease-free survival.
Mutational inactivation of INK4 family members appears to be rare in prostate carcinoma. Komiya and associates (1995) identified a mutation of TP16 in only 1% of clinical specimens. Park and associates (1997) examined primary prostate carcinomas for mutations in TP16, TP15, and TP18 and identified one mutation in TP16. In contrast, inactivation of TP16 by hypermethylation has been implicated in prostate cancer. Herman and associates (1996; 1995) demonstrated hypermethylation of TP16 in 60% of prostate cancer cell lines, whereas TP15 was rarely inactivated. However, these results are tempered by observations that TP16 gene silencing by promoter hypermethylation often occurs during the establishment of cell lines.
Despite the critical role that CIP/KIP family members play in G1S cell cycle arrest, they are rarely mutated in a wide variety of malignancies, including genitourinary tumors, and there are only rare reports of promoter hypermethylation (Shiohara et al, 1994; Kawamata et al, 1995). However, expression of this family of CDK inhibitors plays an important role in cancer in general (Catzavelos et al, 1997; Loda et al, 1997; Yatabe et al, 1998), and in genitourinary carcinomas in particular. Stein and associates (1998) found increased expression of TP21 in 64% of bladder tumors and that increased expression was associated with decreased probability of tumor recurrence and improved patient survival. Decreased TP27 expression has also been linked to increasing tumor grade, pathologic stage, and poor survival in bladder carcinoma (Del Pizzo et al, 1999).
The expression of TP21 in prostate cancer has not demonstrated a clear correlation with advanced disease or poor outcome (Kibel and Isaacs, 2000). However, specific genetic polymorphisms in TP21 and TP27 have been associated with advanced disease (Kibel et al, 2003), and altered expression of TP27 has been implicated in aggressive disease in multiple studies. Cordon-Cardo and colleagues (1998) examined radical prostatectomy specimens and found that absent or low TP27 expression by immunohistochemistry was an independent risk factor for decreased disease-free survival by multivariate analysis. Cote and colleagues (1998) found that decreased TP27 nuclear staining not only correlated with decreased disease-free survival but also overall survival in radical prostatectomy patients, while Freedland and colleagues (2003) found that TP27-positive cells in the prostate needle-biopsy specimen had a 2.5-fold increased risk of biochemical recurrence (prostate-specific antigen [PSA] relapse).
The relevance of TP27 to prostate cancer is also supported by studies of mouse models. For example, mice deficient in TP27 develop prostate hyperplasia, confirming the potential importance of this gene in prostate tissue homeostasis (Cordon-Cardo et al, 1998). More excitingly, studies have shown that mice deficient in both TP27 and Pten have a high incidence of prostate cancer (Di Cristofano et al, 2001).
Cell Cycle Checkpoints
Although beyond the R point the cell cycle functions autonomously, in a unidirectional manner, the cell is still capable of halting its progress at various points should adverse conditions arise. Thus a number of so-called “checkpoints,” first discovered in yeast, have been identified, the two primary ones being the G1S, and G2M checkpoints (Weinert and Hartwell, 1988). These checkpoints are primarily tasked with monitoring the completion of key events in the cell cycle, although genomic integrity is also closely monitored (Hartwell et al, 1994). Loss of checkpoint control can allow inappropriate cell division to occur and also facilitates genetic instability, two key contributors to cancer progression. Indeed, inherited mutations in several of the genes involved in cell cycle checkpoint control (e.g., TP53 and ATM) cause familial cancer predisposition syndromes.
The G1S Checkpoint
As previously mentioned, the expression of cyclin D during G1 is dependent upon the integration of both pro- and antiproliferative signals received from the extracellular environment (e.g., mitogens, nutrient and oxygen availability, etc.); thus the cell will not fully commit to division unless it receives a sustained signal to do so and conditions remain favorable (Matsushime et al, 1991).
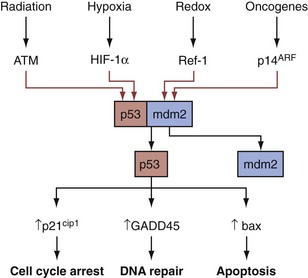
A critical contributor to the G1S checkpoint is the TP53 protein. As discussed elsewhere in more detail, the TP53 tumor suppressor is a pleiotropic transcription factor that receives signals from multiple types of cell stress and damage and, in response, activates genes that function variously in cell cycle arrest, DNA damage repair, and apoptosis (programmed cell death), thus highlighting the link between cell cycle arrest and DNA repair (Elledge, 1996) (Fig. 18–9). DNA damage has obvious relevance for the G1S checkpoint because a normal cell will not proceed to S phase if the genomic template DNA is damaged, thus intact checkpoint controls represent important antitumor mechanisms acting to limit the proliferation of cells bearing potentially oncogenic mutations. During the cellular response to damaged DNA, specific kinases are activated that phosphorylate the TP53 protein, thereby protecting it from HDM2 ubiquitin ligase–mediated degradation (Momand et al, 1992). The accumulated TP53 undergoes further modifications that serve to activate it and facilitate its translocation to the nucleus, where it induces various checkpoint and DNA repair proteins. TP53 target genes that facilitate the G1S checkpoint include TP21 and 14-3-3-sigma, which cause cell cycle arrest by inactivating or sequestering G1 CDK complexes (Giaccia and Kastan, 1998). If the cell cannot arrest growth and/or repair the DNA damage, TP53 often induces apoptosis (Levine, 1997).
S-Phase Arrest
Whereas S phase is not considered to have a checkpoint per se, such as those recognized during G1S and G2M, the cell does maintain the ability to arrest the cell and repair DNA damage through two mechanisms, both mediated through the ATM kinase. In response to genotoxic insults, CDC25A is ubiquitinated through an ATM-dependent cascade, resulting in a rapid decrease in CDC25A levels. Because CDC25 family members normally activate CDK2, the loss of CDC25A function maintains CDK2 in its inactive form, preventing its phosphorylation of substrates required for DNA synthesis. Inactive CDK2 prevents CDC45 from recruiting DNA polymerase. Thus this cascade pauses the cell in S phase, ostensibly to allow time for DNA repair (Bartek et al, 2004).
The second mechanism mediated by ATM in response to DNA damage is phosphorylation of DNA repair enzymes (see DNA Repair). These two parallel pathways further highlight the interaction between DNA repair and checkpoint control. It is not surprising that such S-phase control has been documented in response to DNA-damaging agents, because it is the last opportunity to repair DNA damage before replication (Falck et al, 2002; Pichierri and Rosselli, 2004). Notably, the cascade does not appear to be mediated by TP53 and therefore is an intriguing potential target for therapy in the future (Kastan and Bartek, 2004).
G2M Checkpoint
The G2M checkpoint is a second major point of control during cell division. Unlike the G1S checkpoint, which responds to a variety of extracellular signals in addition to DNA damage, the G2M checkpoint appears to be similar to S-phase arrest and responds only to DNA damage. It therefore serves as an important monitor of DNA replication errors (Weinert, 1997; Bartek et al, 1999).
DNA damage secondary to radiation is believed to induce G2M arrest through ATM. ATM activates CHK1 and CHK2 kinases in response to DNA damage, and these, in turn, inactivate the CDC25 family proteins through phosphorylation, which causes 14-3-3-sigma to bind and sequester it (Hermeking et al, 1997; Sanchez et al, 1997). There are three CDC25 proteins, primarily functioning at the G1M and S-phase checkpoints as outlined previously, whereas CDC25B and CDC25C primarily function at the G2M checkpoint. CDC25B/C phosphorylation disallows their activation of CDC2, therefore inhibiting the cyclin B and cyclin A-CDC2 complexes (Mailand et al, 2002
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


