Molecular alteration
Correlated with low-grade disease (% of cases)
Correlated with high-grade disease (% of cases)
Reference #
FGFR3
40–88
0–36
RAS
0–44
2–65
p53
0–50
42–92
Rb
21–48
25–64
EGFR
24–49
45–71
HER2/neu
0–74
23–72
PI3-Kinase
18–26
5–16
PTEN
3–49
10–94
TSC1 (LOH)
43–68
68–71
mTOR
–
65
Chromosome 9
21 to >50
10–48
In this chapter we seek to present known and emerging molecular markers of UCa. We have opted to present the molecular characteristics of these lesions using a pathway approach, highlighting areas of overlap between low- and high-grade carcinomas and emerging therapies that are pathway-specific in nature.
Fibroblast Growth Factor Receptor 3 (FGFR3)
Low-grade disease is thought to arise from localized urothelial hyperplasia and commonly reported genetic changes involve alterations in the FGFR3, constitutive activation of the HRAS oncogene and loss of heterozygosity of chromosome 9 [6, 10–15]. FGFR3 is a tyrosine kinase receptor that is thought to signal through the RAS signaling pathway (Fig. 33.1) [16–18].
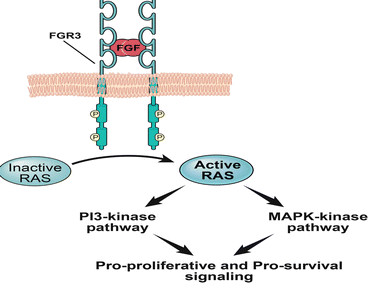
Fig. 33.1
FGFR-3
Fibroblast growth factor receptor 3 (FGFR3) is a tyrosine kinase receptor that is thought to signal through the RAS-MAP-Kinase and PI3-Kinase pathways, predominantly in non-invasive, low-grade disease. Initially, specific gain of function germline mutations in exon 7, 10 and 15 associated with constitutive activation of FGFR3 were linked to autosomal dominant skeletal dysplasia syndromes such as thanatophoric dysplasia [19–27]. Subsequently, similar mutations were identified in UCas [28].
Multiple studies have reported an inverse correlation between the incidence of FGFR3 gene mutations and UCa pathologic stage as well as grade [29, 30]. As both activating mutations in HRAS and FGFR3 are thought to signal through the RAS-mitogen activated protein kinase (MAP-Kinase) pathway, initial studies had hypothesized that both of these signaling pathways were predominantly seen in non-muscle invasive bladder cancers (NMIBC). However, while there is strong evidence for the preferential activation of FGFR3 in low-grade UCa, current studies suggest that HRAS mutations do not exhibit a strong correlation with tumor grade or stage [31– 33]. In fact a recent study suggested that HRAS and FGFR3 mutations appear to be mutually exclusive events [32].
The increased incidence of FGFR3 mutations in non-invasive, low-grade disease has prompted multiple studies evaluating positive FGFR3 mutation status in these lesions as a negative predictor of disease progression, recurrence and disease-specific survival [29, 30, 34, 35]. The results suggest that low-grade lesions characterized by activating mutations of FGFR3 are unlikely to progress to aggressive muscle-invasive disease [29, 36–38].
RAS
Of the three human RAS oncogenes, the Harvey (H)-RAS gene is frequently mutated in bladder cancer [31–33, 39–50]. HRAS mutations in UCa are presumed to contribute to oncogenesis by aberrant activation of the MAP-Kinase and Phosphatidylinositol 3-Kinase (PI3-Kinase) pathways [31–33].
Constitutively active RAS signaling contributes to oncogenesis in bladder cancer by the downstream activation of MAP-Kinase and PI3-Kinase pathways. Activating mutations have been described for HRAS and KRAS, while mutations of NRAS are rare. Initial reports identified point mutations in codon 12, 13 and 61 in a small subset of cases that led to constitutively active HRAS signaling and subsequent studies suggested that point mutations in codon 12 are the most common mutation [32, 39–41]. A single report identified concurrent codon 12 and intron D mutations in high-grade tumors associated with elevated HRAS expression, however the frequency of this event is not clear [50]. There is no clear consensus on the reported frequency of HRAS mutations and association to tumor stage or grade in UCC (Table 33.2).
Table 33.2
RAS mutations in bladder cancer
Year | No. of cases | RAS mutation status | Correlation with tumor grade and stage | Reference |
|---|---|---|---|---|
1985 | 38 | HRAS: 2/38; KRAS: 1/21 | – | Fujita et al. [39] |
1988 | 24 | HRAS: 4/24 | – | Visvanathan et al. [40] |
1990 | 33 | HRAS: 13/33 | None | Czerniak et al. [42] |
1992 | 67 | HRAS: 30/67 | Correlated with high-grade disease | Czerniak et al. [50] |
1993 | 152 | HRAS: 9/152 | None | Knowles et al. [43] |
1993 | 111 | HRAS: 33/111 | – | Levesque et al. [44] |
1994 | 50 | HRAS: 9/50 | None | Burchill et al. [45] |
1994 | 62 Ta/T1 cases; 35 recurrences | HRAS mutation frequency assessed | Increased incidence in low-stage disease | Ooi et al. [46] |
1995 | 100 cases; 19 follow up samples | HRAS: 44/100 | Increased detection of low-grade disease | Fitzgerald et al. [47] |
1995 | 39 | HRAS: 1/39; KRAS: 2/39 | Correlated with high-grade, high-stage disease | Uchida et al. [48] |
1998 | 55 | KRAS: 2/55 | Correlated with high-stage disease | Olderoy et al. [49] |
2001 | 14 | KRAS: 4/14 | – | Ayan et al. [51] |
2005 | 98 | RAS: 13/98 cases; HRAS: 9, KRAS2: 3, NRAS: 2. | None | Jebar et al. [32] |
2009 | 69 primary cases; 23 recurrences | RAS: 11 cases; HRAS: 5, KRAS: 6. | – | Platt et al. [31] |
2010 | 257 primary cases; 184 recurrences from 54 patients | RAS: 28/257 cases; HRAS: 14, KRAS: 14 | None | Kompier et al. [33] |
One study involving 67 tumor samples identified HRAS mutations in over 44 % of cases, with a higher incidence in high-grade disease [50]. In contrast, another study based on a sample size of 152 bladder cancer specimens had a more conservative estimate of a 6 % incidence of HRAS mutations [43]. Moreover, the same study suggested a lack of correlation between tumor grade and/or stage [43]. Mutations have also been described for KRAS2 at a lower frequency than what has been reported for HRAS, while mutations of NRAS are rare [31–33, 48, 49, 51]. More recent studies have estimated the combined frequency of RAS mutations to be between 11 and 13 % (involving a sample size of 92–257 tumor specimens) and have corroborated a lack of correlation between tumor grade and/or stage [31–33].
TP53
Common molecular alterations that have been found to contribute to the development of invasive high-grade UCas commonly include loss of the p53, retinoblastoma (Rb) and phosphatase and tensin homolog (PTEN) tumor suppressor genes as well as loss of heterozygosity events of chromosome 9 [4, 6, 10, 12, 14, 52]. p53, encoded by the TP53 gene on chromosome 17p13.1 behaves like a tumor suppressor by inhibiting the G1-S cell cycle transition [53–55]. Transcriptional activation of the p21 WAF1/CIP1 gene significantly contributes to this regulation by acting as a cyclin-dependent kinase inhibitor (CDKI) (Fig. 33.2) [56, 57].
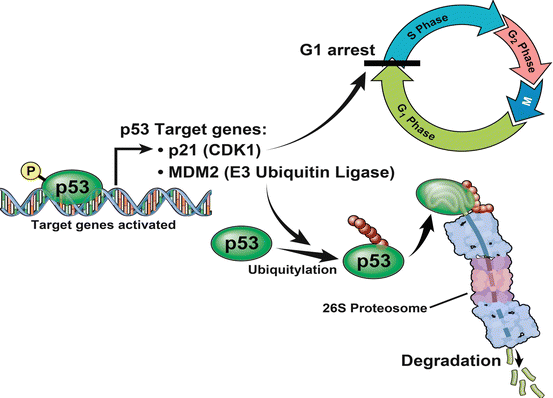
Fig. 33.2
p53
The p53 tumor suppressor inhibits the G1-S cell cycle transition by transcriptionally activating the p21 cyclin-dependent kinase inhibitor (CDKI). MDM2, an E3 Ubiquitin Ligase, is also a transcriptional target of p53 and participates in a negative feedback loop by mediating the proteasomal degradation of p53. Most UCa exhibit loss of heterozygosity at the 17p locus and paradoxically, loss of function mutations in the remaining allele leads to increased protein stability, a prolonged half-life and this facilitates its immunohistochemical detection within the nucleus compared to normal cells where p53 expression is below the threshold of detection by similar techniques [58–60]. Studies have suggested that immunohistochemical detection of p53 can detect mutations accurately in up to 90.3 % of cases, in comparison to more stringent methods such as gene sequencing [58, 61]. Immunohistochemistry based detection of p53 mutation status is limited by false positives, commonly due to the induction of non mutant p53 in response to various stresses and false negatives (due to no detectable protein expression that can be seen in rare instances of homozygous deletions and epigenetic silencing) [58, 62–64].
Clinical assessment of p53 mutations, commonly based on immunostaining results that suggest increased nuclear accumulation, has been correlated with advanced pathological stage and grade of bladder cancers and has also been implicated as a prognostic factor in determining disease progression and recurrence [52, 59, 65–71].
Cell cycle regulation by p53, as mentioned above, is primarily mediated by the transcriptional activation of the p21 WAF1/CIP1 gene, which acts as a cyclin-dependent kinase inhibitor (CDKI) [56, 57]. Loss of p21 (encoded by the p21 WAF1/CIP1 gene), in the presence of p53 mutations has also been reported to be an independent predictor of bladder cancer progression [59, 72]. Other important mediators of p53 signaling include the E3 Ubiquitin Ligase: MDM2 [73]. MDM2 is transcriptionally induced by p53 and participates in a negative feedback loop by mediating the proteasomal degradation of the p53 tumor suppressor (Fig. 33.2) [73]. MDM2, located at the 12q14.3-15 locus, has been shown to be amplified in UCas in a manner that is correlated with high-stage and high-grade disease [59, 74, 75].
Retinoblastoma (Rb)
The retinoblastoma (Rb) gene, located at chromosome 13q14, is recognized as a critical tumor suppressor gene that like p53 also regulates the G1-S cell-cycle transition [59, 76–78]. The E2F-1 transcription factor that is critical to the G1-S transition is sequestered by the active (dephosphorylated) form of Rb, enabling the latter to act as a negative regulator of cell proliferation (Fig. 33.3) [59, 79]. Rb is maintained in its dephosphorylated state by CDKIs such as p16, encoded by the INK4alpha gene at the CDKN2A locus (Fig. 33.3) [59, 80]. Rb phosphorylation, on the other hand, by cyclin dependent kinase (CDK) complexes leads to the release of the E2F transcription factor and begins a program of gene expression that initiates the transition into the S phase of the cell cycle [59, 81, 82]. Rb has been identified as an important tumor suppressor that regulates bladder cancer progression and mutations have been identified predominantly in invasive, late-stage disease [61, 77, 78, 83–86].
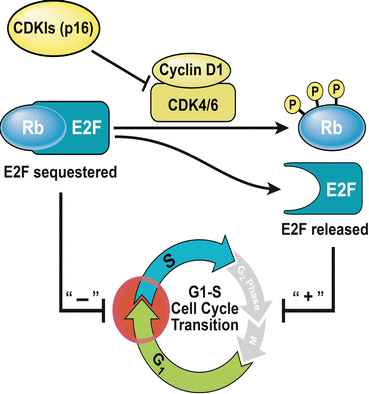
Fig. 33.3
Retinoblastoma (Rb): Sequestration of the E2F-1 transcription factor by the dephosphorylated form of Rb, enables the latter to act as a tumor suppressor by impeding the G1-S transition. Cyclin-dependent kinase inhibitors (CDKI) such p16 also act as tumor suppressors by maintaining Rb in its active, dephosphorylated state. Rb phosphorylation by cyclin dependent kinase (CDK) complexes releases this inhibition, thereby allowing cells to proliferate
Mechanisms of inactivation include deletion events (low Rb expression) as well as hyper-phosphorylation due to positive regulation by CDK complexes or loss of negative regulation by CDKIs such as p16 due to epigenetic silencing (high Rb immunoreactivity, as determined by IHC) [59, 80, 87, 88]. CDK complexes that phosphorylate Rb include cyclin D1 and CDK4 [59]. However, contrary to expectations, increased cyclin D1 expression has been reported to be correlated with positive outcomes in late-stage, muscle-invasive disease while CDK4 overexpression has been linked to high-stage and high-grade disease highlighting the complexity of the underlying regulatory processes [59, 75, 89, 90].
A combination of Rb and p53 along with other relevant markers (MDM2 and CDKIs such as p21, p27 and p16) has been used in multiple studies to successfully predict patient outcomes [59, 91–95]. Various therapeutic regimens that have been targeted towards patients harboring p53 mutations include cisplatin-based regimens that selectively target these cells by inducing apoptosis [59, 96, 97]. Other drugs that target cells with mutant p53 and Rb are in various stages of evaluation for the treatment of UCas. These include the small molecule agent: PRIMA-1, which partially promotes p53 reactivation and nonspecific CDKIs like L86-8275 and UCN-01 [59, 98–100].
The ErbB Family of Receptor Tyrosine Kinases
The ErbB family, commonly implicated in bladder cancer, includes the epidermal growth factor receptor (EGFR; also known as ErbB1and HER1) and HER2/neu (also referred to as ErbB2) (Fig. 33.4).
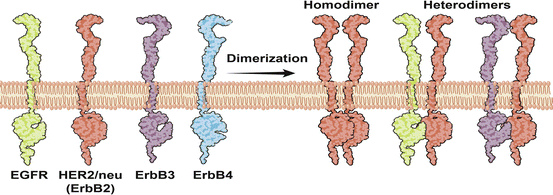
Fig. 33.4
The ErbB family of receptor tyrosine kinases. The ErbB family of receptor tyrosine kinases, commonly implicated in bladder cancer, includes the epidermal growth factor receptor (EGFR; also known as ErbB1and HER1) and HER2/neu (also referred to as ErbB2). The known ligands for EGFR include epidermal growth factor (EGF), amphiregulin (AR), transforming growth factor-α (TGFα), epiregulin (EPR), betacellulin (BTC) and the heparin-binding EGF-like growth factor (HB-EGF), while there are no known ligands for HER2. Activation of EGFR and HER2 involves heterodimerization with other ligand-bound members of the ErbB family of receptor tyrosine kinases and downstream signaling cascades that are activated include the MAP-Kinase and PI3-Kinase pathways
Recent work in this field has sought to address the broader question of the expression pattern of each of the 4 type I tyrosine kinase growth factor receptors (EGFR, ErbB2, ErbB3 and ErbB4) in urothelial carcinomas [101, 102]. Two separate studies have independently suggested that increased EGFR expression appears to be an early event in carcinogenesis and that it is not correlated with tumor grade, stage or survival [101, 102]. It must also be noted there is a considerable amount of debate regarding EGFR immunostaining patterns. There are multiple reports that suggest that EGFR expression is localized to the basal areas of the urothelium supporting the hypothesis that this naturally prevents urinary EGFR-ligands from promoting basal urothelial cell proliferation [103–105]. Contrary to this, Rotterud et al. have reported identifying EGFR expression in all layers of the urothelium suggesting that variability in expression patterns may be attributed to changes in epitope accessibility that might be mediated by lipid rafts [101, 106–108]. Like EGFR, HER2 (ErbB2) also shows increased expression in UCa with no distinct correlation with tumor grade, stage or survival [101, 102, 108–115].
Immunohistochemical detection of membrane localized ErbB3 has not shown a distinct pattern of up or down regulation in UCa and this may possibly be attributed to the presence of a secreted form [101, 116]. Future studies that account for this form will be required to address this question more effectively. On the other hand, multiple studies suggest that ErbB4 is possibly required to maintain the differentiation status of normal urothelium, where high expression levels have been noted as has been reported for multiple tissue types [101, 102, 117].
EGFR (HER1, ErbB1)
Initial studies based on competitive binding analysis and immunohistochemistry suggested that EGFR was overexpressed in bladder cancer with preferential expression in advanced, late-stage disease in studies that involved sample sizes ranging from 21 to 173 cases [118–121].
The known ligands for EGFR include epidermal growth factor (EGF), amphiregulin (AR), transforming growth factor-α (TGFα), epiregulin (EPR), betacellulin (BTC) and the heparin-binding EGF-like growth factor (HB-EGF) (Fig. 33.4) [122]. In a study aimed at determining relative EGFR-ligand expression, EGF was reported to be expressed at very low levels leading to the hypothesis that it might function in an endocrine fashion as opposed to other more abundantly expressed EGFR ligands that possibly had an autocrine or paracrine mechanism of action [123, 124]. HB-EGF, EPR and TGFα, in particular, were found to be expressed at much higher levels and this was correlated with advanced pathological stage and poor patient survival [123, 124]. However, even though EPR expression had the strongest correlation with patient survival, in vitro studies suggest that EPR has a much lower binding affinity for EGFR compared to EGF in bladder cancer cells, raising the possibility that it might signal in a noncanonical fashion through alternate receptors [123, 125].
EGFR mediates its actions by heterodimerization with other ligand-bound members of the ErbB family of receptor tyrosine kinases and downstream signaling cascades that are activated include the MAP-Kinase and PI3-Kinase pathways (Fig. 33.4) [114, 126, 127].
For instance, in vitro studies suggest that EGFR promotes invasive behavior of bladder cancer cells through both RAS-dependent (MAP-Kinase pathway) as well as in a RAS-independent, PI3-Kinase facilitated manner [127]. Similarly, urothelium-specific overexpression of either EGFR or the downstream HRAS oncogene (associated with increased activation of the MAP-Kinase pathway) in transgenic mice led to urothelial hyperplasia phenotype [128, 129]. Double transgenic mice overexpressing both EGFR and the downstream HRAS oncogene did not exhibit a tumor-enhancing effect, suggesting pathway redundancy [128]. This was in contrast to double transgenic mice that overexpressed EGFR and had a functional inactivation of the retinoblastoma and p53 tumor suppressor genes [due to Simian virus 40 (SV40) large T antigen expression] that led to high-grade bladder carcinomas [128]. This suggests that increased EGFR signaling coupled with non-redundant genetic events can have a synergistic effect, contributing to bladder cancer progression.
Promising EGFR-tyrosine kinase inhibitors include 4,5-Dianilinophthalimides as well as the monoclonal antibody cetuximab [130, 131]. These have been found to be effective in reducing the orthotopic tumor burden in mice, partially attributed (in the case of cetuximab) to reduced angiogenesis [130, 131]. In fact a phase II trial involving a combination of cetuximab with standard chemotherapeutic regimens is currently underway [132]. Challenges to cetuximab therapy include acquired resistance to this drug, which according to one report has been linked to increased phosphorylation of the C-terminal fragment of HER2 in vitro [133]. In one study, a panel of molecular markers characterized by increased expression of ErbB4 and E-cadherin, suggestive of a well-differentiated urothelial identity, predicted increased sensitivity to cetuximab [134]. From a therapeutic perspective, this calls for the comprehensive evaluation of biomarkers that would predict responsiveness to cetuximab as well as the trial of combination therapy with dual EGFR/HER2 kinase inhibitors [133].
Additional tyrosine kinase inhibitors that are being evaluated for therapeutic purposes in bladder cancer include Gefitinib (“iressa”, ZD1839), which may possibly also inhibit EGFR/ErbB2 and EGFR/ErbB3 heterodimers [135, 136]. Preliminary studies suggest that responsiveness to this class of drugs does not necessarily correlate with EGFR protein levels [137]. In contrast to its in vitro efficacy, attributed to inhibition of MAP-Kinase and PI3-Kinase signaling, it has been shown to be effective in only certain cell types in mouse-xenograft studies for reasons that are not clearly understood [135]. In a carcinogen-induced model of urinary bladder cancers (in rats) Gefitinib was found to increase tumor latency, reduce tumor incidence as well as reduce tumor volumes in a dose dependent manner [138]. The results of this study were attributed to reduced VEGF dependent angiogenesis and a down regulation of cell-cycle genes related to the anaphase protein complex, in addition to inhibiting EGFR signaling [138]. Therefore, dual vascular endothelial growth factor receptor (VEGFR)/EGFR antagonists such as vandetanib (ZD6474) have attracted much interest due to their ability to suppress both angiogenesis and cell proliferation [139, 140]. Preliminary in vitro studies with vandetanib have shown promising results and this warrants further investigation [139, 140].
HER2/neu (ErbB2) been reported to show increased expression in UCa, with conflicting evidence regarding correlation with tumor grade, stage and survival [101, 102, 108–114, 141–143]. Some studies have shown a correlation with worsened pathological stage while its potential role as a prognostic variable is more controversial [101, 108, 110, 113, 115, 144]. One study (involving 73 cases) interrogated the relative abundance of ErbB receptor tyrosine kinase expression that might contribute to bladder cancer oncogenesis and progression, at the level of mRNA transcript expression [123]. The results suggested that while HER2 was markedly more abundant than EGFR, neither was correlated with patient survival and the same conclusion has been supported by multiple studies [101, 102, 121, 123, 145–147].
While HER2 gene amplification and protein expression levels have been reported to show a high degree of correlation in the rarer and highly aggressive UCa variants: micropapillary carcinoma, this correlation is not seen in conventional UCa [148]. HER2 gene amplification rates have been reported to be much lower (7–9 % of cases) than corresponding protein expression levels (where measured in tandem) leading to the conclusion that HER2 is significantly regulated through either transcriptional or post-transcriptional mechanisms that are yet to be elucidated in the context of bladder cancers [111, 149, 150]. A recent retrospective study that assessed over 1,000 patients estimated high HER2 protein expression in 9.2 % of cases with associated gene amplification in only 5.1 % of these [151].
Jiminez et al. [109] reported a higher HER2 expression in paired lymph nodes when compared to the corresponding primary tumors suggesting a significant role of HER2 in the metastatic process. However, these findings were not corroborated in future studies [101]. Interestingly, multiple reports have since demonstrated significantly lower HER2 expression in metastatic lesions with increasing distance from the primary tumor suggesting that alternate signaling pathways may be more relevant at these sites [112, 152].
Though there are no known ligands for HER2, it is thought to be activated by heterodimerization with other ligand-bound members of the ErbB family of receptor tyrosine kinases and downstream effectors include MAP-Kinase, PI3-Kinase signaling cascades as well as MYC [153, 154]. Interestingly, MYC and the TOP2A gene (the latter codes for DNA topoisomerase 2-alpha) have been reported to be co-amplified with HER2 [114, 155, 156]. In one report, MYC was co-amplified with HER2 in 57 % cases, all of which occurred only in the metastatic population associated with locally advanced high-stage (pT4) disease [114]. This is significant as HER2 and MYC co-amplification has been found to influence HER2-directed therapy in other cancer types [155, 157].
Despite the inconclusive data regarding the prognostic value of HER2 as a marker of tumor progression, bladder cancer is an entity with one of the highest levels of HER2 expression [4, 158]. This has prompted an evaluation of HER2-directed therapeutic agents such as trastuzumab (a humanized monoclonal antibody directed against HER2) and lapatinib (a dual EGFR/HER2 tyrosine kinase inhibitor) [108, 159]. HER2-directed therapy has been shown to provide maximal benefit in the presence of gene amplification. Immunohistochemical detection of HER2 expression has therefore been proposed as a screening tool to identify cases of HER2 gene amplification using fluorescent in situ hybridization (FISH) [114, 160].
Initial trials of trastuzumab in combination with standard chemotherapeutic agents in small trials of 6–7 patients with HER2 over-expressing metastatic or locally advanced disease suggested that the drug was well tolerated but did not offer a clear trend with regards to the therapeutic response [161, 162]. In a more extensive phase II trial, combination therapy with trastuzumab in a group of 44 patients with HER2 positive tumors had a promising overall response rate of 70 % [163]. On the other hand, a phase II trial of lapatinib monotherapy in 34 patients showed limited benefit that was found to be correlated with EGFR status and to a lesser extent HER2 expression [164]. Further studies are required to identify the molecular signatures of tumors that are likely to respond to these therapeutic interventions.
PI3-Kinase Signaling Cascade
Class IA PI3-Kinases are composed of a regulatory subunit (p85-alpha, p85-beta and p55-gamma encoded by PIK3R1, PIK3R2 and PIK3R3, respectively) and a catalytic subunit (p110-alpha, p110-beta and p110-delta encoded by the PIK3CA, PIK3CB and PIK3CD genes) [165–168]. Of these, PIK3CA is commonly mutated in bladder cancers [31, 33, 169]. Growth factors (signaling through receptor tyrosine kinases), activated RAS as well as G-protein coupled receptors can activate the catalytic subunit of PI3-Kinase leading to the phosphorylation of phosphatidylinositol bisphosphate (PIP2) to phosphatidylinositol triphosphate (PIP3) [167, 168, 170–174]. This leads to the recruitment of AKT1 to the cell membrane where it is activated by a series of phosphorylation events by phosphoinositide-dependent kinase 1 (PDK1) and the mammalian target of rapamycin complex 2 (mTORC2) [168, 175–184].
AKT, in particular, has been referred to as a “master” kinase that regulates numerous pro-survival signaling cascades implicated in cancer progression [185]. Downstream signaling mediators of AKT that have been shown to be significant for bladder cancer progression include the mammalian target of rapamycin complex 1 (mTORC1) [178].
PI3-Kinase
The incidence of mutations in the catalytic component of PI3-Kinases (PIK3CA) has been estimated to be as high as 24–25 % in studies that assessed primary tumors from up to 257 patients as well as recurrences where the sample size ranged from 23 to 54 patients [31, 33]. Common mutations in PIK3CA have been found to be localized to the helical and kinase domains and induce gain of function through distinct mechanisms, that vary in their dependence on either the inhibitory effect of the p85 regulatory subunit or their interaction with RAS [31, 186].
Mutations in PIK3CA appear to have a higher incidence in low-grade and low-stage disease but were not found to be accurate predictors of recurrence-free, progression-free or disease-specific survival [33, 169]. The above-mentioned study also reported a high co-occurrence of mutations in FGFR3 (RAS-MAPK activation) with mutations in the PIK3CA gene [33]. This is particularly significant as receptor tyrosine kinase targeted therapies (EGFR targeted therapies in clinical trials: CALBG-90102, NCT00088946, NCT00380029) are likely to have limited efficacy in the presence of downstream PIK3CA mutations [33].
PTEN
The phosphatase and tensin homolog (PTEN), through its lipid phosphatase activity catalyzes PIP3 to PIP2, thereby preventing activation of AKT1 downstream of PI3-Kinase [182, 187]. Loss of the PTEN tumor suppressor gene at chromosome 10q23 was found to predispose to Cowden disease, an autosomal dominant cancer predisposition syndrome characterized by occasional cases of bladder cancer [84]. Initial studies estimated the incidence of loss of heterozygosity (LOH) events at the 10q locus at about 6.6 % in superficial (non-muscle invasive) urothelial cancers and up to 24.5 % of cases of late-stage, muscle-invasive bladder cancer [31, 84, 188, 189]. However, these and subsequent studies have shown a low incidence of biallelic inactivation of PTEN suggesting that loss of the PTEN tumor suppressor by itself is unlikely to be the primary regulator of bladder cancer progression [84, 188, 190].
Nevertheless, loss of PTEN has been found to be correlated with disease stage and grade [187, 188, 191]. In one study, PTEN expression was found to be significantly reduced in 94 % of cases representing late-stage disease in contrast to only 42 and 8 % of superficial papillary urothelial carcinomas and carcinomas in situ, respectively [187]. A developing focus of ongoing research has been directed at noncanonical functions of PTEN in promoting cancer progression that involve its subcellular localization. Nuclear localization of wild-type PTEN has been found to be critical for maintaining centromere stability and the downregulation of various cell cycle factors such as cyclin D1 that eventually induce a G0-G1 arrest [178, 192–194]. In fact, a new study of 63 primary bladder cancer patients reported a significant reduction in the nuclear expression of PTEN in advanced, late-stage disease [187]. A subsequent study involving 69 primary and 23 recurrent bladder cancer samples corroborated the finding of reduced expression of PTEN (49 % of cases), with a predominance of reduced nuclear expression [31].
A combination of reduced PTEN mRNA expression along with specific mutations of PIK3CA, with or without amplification of the HER2 gene has been proposed to be a good predictive tool for determining PI3-Kinase pathway activation [195]. Indeed, in an analysis of 80 bladder cancer samples, this PI3-Kinase pathway activation signature was strongly correlated with patient outcomes based on overall survival [190, 195].
Furthermore, increased activation of the PI3-Kinase pathway in the urothelium of transgenic mouse models has been shown to contribute to urothelial hyperplasia [128, 129] [187]. Specifically, overexpression of either EGFR or the downstream HRAS oncogene, that was spatially restricted to the urothelium, led to an urothelial hyperplasia phenotype while a deletion of PTEN within the urothelium led to the spontaneous development of papillary urothelial carcinomas in at least 10 % of the mice studied [128, 129, 187]. Additionally, in the latter case, the frequency of papillary urothelial carcinomas was found to be significantly increased following exposure to a chemical carcinogen and the resultant lesions showed a strong correlation with increased AKT activation, a hallmark of PI3-Kinase signaling [187].
Mechanistic studies have also shown that decreased signaling through the PI3-Kinase pathway, as a consequence of PTEN overexpression, is correlated with decreased cancer-cell invasiveness in both in vivo orthotopic assays as well as in vitro organotypic assays [196]. The same study showed that PTEN can inhibit tumor invasion even in the absence of its lipid phosphatase activity and suggested a role for its protein phosphatase activity, which is poorly understood [196].
p53 and PTEN
Recent studies by Puzio-Kuter et al. evaluated the effect of monoallelic and biallelic, urothelium-specific deletions of the p53, Rb and PTEN tumor suppressor genes alone, or in combination on bladder cancer progression [197]. The simultaneous inactivation of both p53 and PTEN led to the spontaneous development of a wide spectrum of invasive lesions, characterized by increased downstream signaling through mTORC1 [197]. Consistent with these results, this study also reported that a high percentage of late-stage, muscle-invasive patient samples that had altered p53 also had deregulated PTEN expression [197]. Future studies involving larger datasets will be required to determine the relative frequency of combined PTEN inactivation and p53 alterations and to identify effective therapeutic targets that counter these molecular changes.
Wnt Signaling and PTEN
Unlike colorectal cancers, where deregulated Wnt signaling due to somatic mutations in the adenomatous polyposis coli (APC) gene has been commonly described, its role in bladder cancer is controversial [198–201]. A number of studies have suggested increased activation of Wnt signaling in bladder cancer, based on epigenetic silencing of secreted receptor protein antagonists (such as the Wnt-inhibitory factor-1), loss of function mutations and deletions of the APC tumor suppressor gene and immunohistochemical detection of the key signaling intermediate: β-catenin [201–210]. Recent studies in mouse models have suggested an important interaction between the Wnt signaling pathway and the PTEN tumor suppressor [201]. In these studies, urothelium-restricted over activation of Wnt signaling led to hyper-proliferative lesions that did not undergo malignant transformations and were characterized by increased PTEN expression [201]. However, increased Wnt signaling in a PTEN null background promoted a transition to papillary UCas in these mice [201]. This study also reported a correlation between increased Wnt signaling and robust PI3-Kinase pathway activity (due to a loss of PTEN) in a limited number of human bladder cancer cases and therefore calls for further investigation into the incidence of combined dysfunctions in the Wnt and PI3-Kinase signaling pathways in bladder cancer [201].
mTOR
The complex mTORC1 consists of mTOR, mLST8 and regulatory associated protein of mTOR (RAPTOR) [178, 180]. Activation of AKT, as part of PI3-Kinase signaling, leads to a disruption of the tuberous sclerosis complex (TSC) and this releases the inhibition of RAS homolog enriched in brain (Rheb), an activator of mTORC1. Known targets of activated mTORC1 kinase include p70 S6 kinase 1 (S6K1) (which phosphorylates ribosomal protein S6) and eukaryotic initiation factor 4E binding protein 1 (4EBP1) which play key roles in the translation of 5′ terminal oligopyrimidine tract mRNAs and in cap dependent translation initiation, both processes that are critical to the translation of cell cycle regulators such as cyclin D1 [178, 180, 181, 184, 211–214].
In an exhaustive study, involving 803 cases of bladder cancer and 803 controls that were matched for gender and age, single-nucleotide polymorphisms (SNPs) in 19 genes belonging to the PI3Kinase-AKT-mTOR signaling cascade were evaluated as predictors for bladder cancer susceptibility [215]. Germ-line genetic variations in the gene coding for RAPTOR, an mTORC1 component, were found to be correlated with increased risk of bladder cancer along with a significant gene-dosage effect implying that deregulated mTORC1 signaling significantly contributes to bladder cancer risk [215]. Furthermore, Hansel et al. reported a significant correlation between increased mTOR signaling (55–74 % of cases), increased pathological stage and reduced disease-specific survival in an analysis of 121 cases of late-stage, muscle-invasive UCas [216]. This has been corroborated by a subsequent study which looked at multiple parameters corresponding to the PI3-Kinase signaling cascade, including mTORC1 targets, in a broader subset of 887 cases and reported an association between deregulated PI3-Kinase signaling and high-grade, high-stage cases of bladder cancer [191].
There is considerable interest in the mTOR signaling pathway, due to the availability of numerous small molecule inhibitors such as rapamycin and its structural derivatives (temsirolimus, Wyeth pharmaceuticals; everolimus or RAD 001, Novartis pharmaceuticals; deforolimus or AP-23573, Ariad pharmaceuticals) [178, 179]. Rapamycin and its derivatives complex with the immunophilin: FKBP12 so as to primarily inhibit the kinase activity of mTORC1 [178, 179]. While these drugs are being investigated in phase I and II clinical trials for cancers such as metastatic renal cell carcinoma, they have not been evaluated as potential therapeutic agents in the management of UCa, as yet [178, 179, 217]. However, a number of in vitro studies have shown that this class of drugs significantly inhibits mTOR signaling in a dose dependent manner and consequently leads to a G0-G1 cell-cycle arrest, as well as a reduction in VEGF-dependent angiogenesis [216, 218, 219]. The anti-proliferative efficacy of these drugs has also been demonstrated in vivo in mouse xenograft studies [197, 201, 216, 219].
Given the published data on the safety and efficacy of rapamycin derivatives in various cancers, and the increased volume of literature regarding its efficacy in vitro and in multiple mouse models, it is likely that these drugs will soon be evaluated for their therapeutic efficacy in cisplatin-refractory UCa [179].
Chromosomal Abnormalities
In low-grade disease, alterations of chromosome 9 such as loss of heterozygosity are a frequent event and are seen at a similar frequency in late-stage, muscle-invasive disease [4]. This suggested the presence of a common tumor suppressor gene, critical to bladder cancer initiation on chromosome 9 and led to the identification of common areas of loss of heterozygosity (LOH) on both 9p and 9q (Fig. 33.5) [4, 220–225].

Fig. 33.5
Chromosome 9 alterations. Common areas of loss of heterozygosity have been identified on both chromosome 9p and 9q and this is thought to be an important event in bladder cancer initiation. The CDKN2A locus on 9p encodes for the tumor suppressors p16 and p14, which act by regulating p53 and Rb function. Important tumor suppressors that have been mapped to mapped to 9q3 include deleted in bladder cancer chromosome region 1 (DBCCR1) and Tuberous Sclerosis 1 (TSC1)
Chromosome 9p
Mapping studies led to the identification of two regions of LOH on 9p21 [220, 222–224]. As deletion and methylation events were more common at the CDKN2A locus than the CDKN2B locus at this site, the former has been hypothesized to be the primary target of loss of 9p21 [222, 226]. The INK4alpha gene at the CDKN2A locus encodes for the tumor suppressor p16, which regulates Rb so as to inhibit the G1-S transition [59]. Additionally, the alternate reading frame (ARF) gene at the CDKN2A locus encodes for p14, which indirectly regulates expression of the p53 tumor suppressor by interacting with the MDM2 E3 ubiquitin ligase [59]. In one report, homozygous deletions of p16 and p14 leading to deregulated expression of both the Rb and p53 tumor suppressors, respectively, was correlated with poor patient outcomes [227]. p53 and Rb regulation, in the context of bladder cancer development, has been discussed in greater detail earlier in this chapter.
In a direct clinical application, fluorescence in situ hybridization based diagnostics tests (UroVysionTM, Vysis Inc.) are used to detect homozygous deletions at the 9p21 locus [228]. These tests have been used to predict the risk of tumor recurrence for superficial non-invasive lesions, post-therapy [59, 228].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:
 Diet and GU Cancers
Diet and GU Cancers
 Life After Urological Cancer – Psychological Issues
Life After Urological Cancer – Psychological Issues
 Genetics and Genito-Urinary Cancer
Castrate Resistant Prostate Cancer: Systemic Chemotherapy and a System Problem
Genetics and Genito-Urinary Cancer
Castrate Resistant Prostate Cancer: Systemic Chemotherapy and a System Problem
 Renal Failure, Dialysis and Transplantation: In the Management of Renal Cell Carcinoma (Solitary Kidney and Bilateral Renal Tumours)
Renal Failure, Dialysis and Transplantation: In the Management of Renal Cell Carcinoma (Solitary Kidney and Bilateral Renal Tumours)
 High Intensity Focused Ultrasound (Hifu) in Prostate Cancer
High Intensity Focused Ultrasound (Hifu) in Prostate Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


