Chapter 22 Management of Pituitary, Adrenal, and Thyroid Disease
PITUITARY DISORDERS
Anatomy
The optic chiasma may be in front of (15%), above (80%), or behind the sella (5%). Within this optic chiasma, nerve fibers from the nasal half of the retina cross over to the opposite optic tract while those from the temporal half remain uncrossed. The close association of the pituitary gland with the optic chiasma explains the visual symptoms associated with expanding masses in this region.1
The median eminence is an intensely vascular component at the baseline of the hypothalamus that forms the floor of the third ventricle. The pituitary stalk arises from the median eminence. The hypothalamus extends anteriorly to the optic chiasma and posteriorly to the mammillary bodies. It was not until the mid-1960s that hypothalamic releasing hormones were isolated and identified (Table 22-1).
Table 22-1 Pituitary Hormones, Hypothalamic Hormones, and Other Regulatory Factors
| Pituitary Hormones | Hypothalamic Hormones | Other Regulatory Factors |
|---|---|---|
| Thyrotropin | TRH | T4, T3, dopamine, Pit-1 |
| Corticotropin | CRH | ADH, adrenaline, cortisol |
| Luteinizing hormone | LH-RH | Estrogen, progesterone, testosterone |
| Follicle-stimulating hormone | LH-RH | Activin, estrogen, inhibin, follistatin, testosterone |
| Growth hormone | GH-RH | Somatostatin, estrogens, T4, Pit-1 |
| Prolactin | PRF | Dopamine, TRH, Pit-1, estrogen, serotonin, vasoactive intestinal peptide, GnRH-associated peptide |
TRH, thyrotropin-releasing hormone; CRH, corticotropin-releasing hormone;
LH-RH, luteinizing hormone-releasing hormone; GH-RH, growth hormone-releasing hormone; PRF, prolactin-releasing factor; Pit-1, pituitary-specific transcription factor; ADH, antidiuretic hormone.
Pituitary Tumors
Pituitary tumors may present with either hypofunction or hyperfunction, as well as symptoms directly related to the mass effect of the tumor (Table 22-2). Since the advent of computed tomography (CT), microadenomas have been arbitrarily designated as equal to or less than 10mm in diameter and macroadenomas as greater than 10mm in diameter. They are invariably benign, with no sex predilection. Pituitary adenomas are rarely associated with parathyroid and pancreatic hyperplasia or neoplasia as part of the multiple endocrine neoplasia type I (MEN I) syndrome. Pituitary carcinomas are rare, but metastases from other solid malignancies can occur more frequently.2
Table 22-2 Clinical Manifestations of Pituitary Tumors
| Endocrine Effects | ||
|---|---|---|
| Mass Effects | Hyperpituitarism | Hypopituitarism |
| Headaches | GH: acromegaly | GH: short stature in children, increased fat mass, decreased strength and well-being in adults |
| Chiasmal syndrome | Prolactin: hyperprolactinemia | Prolactin: failure of postpartum lactation |
| Hypothalamic syndrome | Corticotropin: Cushing’s disease Nelson’s syndrome | Corticotropin: hypocortisolism |
| Disturbances of thirst, appetite, satiety, sleep, and temperature regulation | LH/FSH: gonadal dysfunction or silent α-subunit secretion | LH or FSH: hypogonadism |
| Diabetes insipidus | Thyrotropin hyperthyroidism | Thyrotropin: hypothyroidism |
| SIADH | ||
| Obstructive hydrocephalus | ||
| Cranial nerves III, IV, V1, V2, and VI dysfunction | ||
| Temporal lobe dysfunction | ||
| Nasopharyngeal mass | ||
| CSF rhinorrhea | ||
SIADH = syndrome of inappropriate antidiuretic hormone; GH = growth hormone; LH = luteinizing hormone; FSH = follicle-stimulating hormone.
PITUITARY ADENOMAS
Approximately 50% of pituitary adenomas are prolactinomas, 15% are growth hormone (GH)-producing, 10% are corticotropin-producing, and less than 1% secrete thyrotropin. Nonfunctioning pituitary adenomas, or more appropriately named nonsecretory adenomas represent about 25% of pituitary tumors. Most of these adenomas on morphologic examination reveal granules containing hormones, typically components of glycoprotein hormones. Autopsy studies suggest that up to 20% of normal individuals harbor incidental pituitary microadenomas that are pathologically similar in distribution to those that present clinically.3
Prolactinoma
Hyperprolactinemia
Hyperprolactinemia impairs pulsatile gonadotropin (luteinizing hormone [LH] and follicle-stimulating hormone [FSH]) release, likely through alteration in hypothalamic luteinizing hormone-releasing hormone (LHRH) secretion. Women of reproductive age usually present with oligomenorrhea, amenorrhea, galactorrhea, and infertility. Those with longstanding amenorrhea are less likely to have galactorrhea secondary to longstanding estrogen deficiency. Postmenopausal women and men usually come to medical attention because of a mass effect, such as headaches and visual field defects.4
Drug history is an important part of the initial evaluation of patients with elevated prolactin level, because some medications are associated with hyperprolactinemia and their discontinuation (if possible) will avoid any further, often expensive, workup. Other common conditions associated with elevated prolactin levels include pregnancy and hypothyroidism (Table 22-3).
Table 22-3 Differential Diagnosis of Hyperprolactinemia
| Physiologic | Pathologic | Pharmacologic |
|---|---|---|
| Pregnancy | Prolactinoma | TRH |
| Postpartum | Acromegaly (25%) | Psychotropic medications |
| Newborn | Hypothalamic disorders | Phenothiazines |
| Stress | “Chiari-Frommel” | Reserpine |
| Hypoglycemia | Craniopharyngioma | Methyldopa |
| Sleep | Metastatic disease | Estrogen therapy |
| Postprandial hypoglycemia | Pituitary stalk secretion or compression | Metoclopramide, cimetidine (especially intravenous) |
| Intercourse | Hypothyroidism | Opiates |
| Nipple stimulation | Renal failure | Verapamil |
| Liver disease | Some SSRIs, including fluoxetine and fluvoxamine | |
| Chest wall trauma (burns, shingles) |
SSRI = Selective serotonin reuptake inhibitor.
The prolactin level usually correlates well with the size of the tumor. Serum prolactin level above 200μg/L is almost always indicative of a prolactin-producing pituitary tumor. However, a serum prolactin level below 200μg/L can be seen in the presence of a large pituitary adenoma, because stalk compression from an adenoma that does not secrete prolactin can also cause hyperprolactinemia. Stimulatory tests, including the TRH stimulation test, which are performed to determine whether an elevated prolactin is a result of a prolactinoma, are nonspecific and cannot be used to diagnose or exclude a tumor. Large prolactinomas may be associated with a falsely low prolactin level. Dilution of serum will reveal the markedly elevated prolactin levels.
Observational studies in patients with microadenomas indicate that serum prolactin concentration or adenoma size increases in only a minority of patients; indeed, serum prolactin deceases in a majority of patients over time. Details of the relationship between prolactin adenomas and amenorrhea are found in Chapter 16.
Treatment
Medical therapy during pregnancy often stirs debate about the continuation of bromocriptine. Tumor-related complications are seen in about 15% of pregnancies and in only 5% of women with microadenomas. A sensible approach would be to stop bromocriptine when pregnancy begins, and then follow the clinical status with serum prolactin levels and visual field examinations. If there is significant worsening in clinical status, bromocriptine could be reinstituted. The adenoma can be followed yearly by MRI, increasing the duration between imaging studies if size is stable.5
Acromegaly
Acromegaly may occur at a rate of 3 to 4 cases per million per year, with mean age at diagnosis of 40 years in men and 45 years in women. The GH-secreting tumors tend to be more aggressive in younger patients. Classical clinical features are listed in Table 22-4. More than 95% of cases of acromegaly are caused by GH-secreting pituitary tumors. In rare cases, they are caused by ectopic GH-releasing hormone (GH-RH) secretion, mainly carcinoids and pancreatic islet cell tumors. Patients with acromegaly have a 3.5-fold increased mortality rate, with cardiovascular disease being the most common cause of death. Somatotrope adenomas appear to be monoclonal in origin. A gsp mutation in a GspIa subunit in GH cells, leading to continuous GH secretion, has been shown to cause acromegaly.6
Table 22-4 Clinical Signs and Symptoms of Acromegaly
| Coarsening of facial features |
| Prominent jaw and frontal sinus |
| Broadening of hands and feet |
| Hyperhidrosis |
| Macroglossia |
| Signs of hypopituitarism |
| Diabetes mellitus (10%–25%) |
| Skin tags (screening for colonic polyps required) |
| Hypertension (25%–30%) |
| Cardiomyopathy (50%–80%) |
| Carpal tunnel syndrome |
| Sleep apnea (5%) |
Insulin-like growth factor-I has a longer plasma half-life than GH and is an excellent initial screening test for those suspected to have acromegaly. An elevated IGF-I level in a clinical setting suggestive of acromegaly almost always confirms the diagnosis. Patients with poorly controlled diabetes and malnutrition may have falsely low serum IGF-I levels. The oral glucose tolerance test remains the gold standard test to confirm the diagnosis. Normal individuals suppress their GH level to less than 1μg/L (using chemiluminescent assays) within 2 hours after ingestion of 100 grams of oral glucose solution.
Treatment
Long-term observations of patients on somatostatin analogues have shown no evidence for tachyphylaxis. Some degree of tumor shrinkage is expected in up to 50% of patients, although in most cases there is less than 50% shrinkage in tumor size. The most common side effects are gastrointestinal, including diarrhea, abdominal pain, and nausea. The most serious side effect of somatostatin analogues is cholelithiasis, seen in up to 25% of patients. Its long-term management is similar to that for cholelithiasis in the general population, and routine ultrasonographic screening is not indicated. There have been very few reports of the use of a somatostatin analogue during pregnancy.7,8
Cushing’s Disease
Corticotropin-secreting pituitary adenoma is the most common cause of endogenous Cushing’s syndrome (60%), with the rest being adrenal (25%) or ectopic (15%) in origin. The term Cushing’s disease refers specifically to a pituitary tumor as the cause. Signs and symptoms suggestive of hypercortisolism are listed in Table 22-5. Many signs and symptoms of Cushing’s disease are nonspecific, including hypertension, abnormal glucose tolerance, menstrual irregularities, and psychiatric disturbances, including depression. Most women with Cushing’s disease have reduced fertility.9
Table 22-5 Signs and Symptoms of Cushing’s Syndrome
| Clinical Feature | Approximate Prevalence (%) |
|---|---|
| Obesity | |
| General | 80–95 |
| Truncal | 45–80 |
| Hypertension | 75–90 |
| Menstrual disorders | 75–95 |
| Osteopenia | 75–85 |
| Facial plethora | 70–90 |
| Hirsutism | 70–80 |
| Impotence/decreased libido | 65–95 |
| Neuropsychiatric symptoms | 60–95 |
| Striae | 50–70 |
| Glucose intolerance | 40–90 |
| Weakness | 30–90 |
| Bruising | 30–70 |
| Kidney stones | 15–20 |
| Headache | 10–50 |
Women with Cushing’s disease typically have fine facial lanugo hair and may have acne and temporal scalp hair loss secondary to increased adrenal androgen secretion. There is usually a 3- to 6-year delay in diagnosis of patients with Cushing’s disease, and it may be possible to date the onset of the disease by determining which scars are pigmented due to excess secretion of corticotropin and other melanotropins.
Diagnosis
Twenty-four hour urinary free cortisol measurement is the single best test for diagnosis of Cushing’s syndrome (Fig. 22-1). Because of the significant overlap between normal individuals and those with Cushing’s syndrome, random serum cortisol has no role in the diagnosis of Cushing’s syndrome. A 1-mg overnight dexamethasone suppression test with a morning cortisol level below 1.8μg/dL virtually rules out the disease but is associated with an up to 40% false-positive rate.
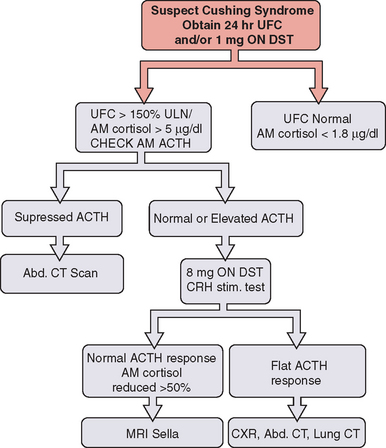
A combination of low-dose dexamethasone suppression test and corticotropin-releasing hormone (CRH) stimulation test has been shown to have 100% diagnostic accuracy in a National Institutes of Health study. This test may have a significant value in establishing the diagnosis in those with pseudo-Cushing’s and elevated 24-hour urinary free cortisol. Other tests useful in establishing the diagnosis of Cushing’s disease include midnight serum and salivary cortisol (see Fig. 22-1).
Once the diagnosis of Cushing’s syndrome has been established, the next step is to find out whether cortisol hypersecretion is corticotropin dependent (see Fig. 22-1). Although an undetectable or low corticotropin level is consistent with adrenal etiology, low-normal corticotropin may be seen in both ectopic Cushing’s syndrome and corticotropin-secreting pituitary tumor. The CRH stimulation test is used to differentiate between the two. Although corticotropin levels tend to be higher in those with ectopic Cushing’s syndrome compared to patients with pituitary disease, there is considerable overlap. The high-dose dexamethasone test or the CRH stimulation test is helpful in differentiation of the two disorders. Cortisol levels are not suppressed with the high-dose (8 mg) dexamethasone test in patients with ectopic corticotropin syndrome, and CRH stimulation may not lead to a further rise in corticotropin.10 The gold standard test to differentiate pituitary Cushing’s syndrome from ectopic corticotropin-producing tumor is inferior petrosal sinus sampling. This test should be performed by an experienced neuroradiologist; it is essential to note that it cannot be used to make the diagnosis of Cushing’s syndrome.
Nonsecretory and Glycoprotein-Secreting Pituitary Adenomas
Glycoprotein-secreting Adenomas
Rarely, pituitary adenomas can secret glycoproteins, including FSH, LH, or thyrotropin. An FSH adenoma may cause amenorrhea in a woman, or an LH adenoma may cause precocious puberty in a boy. Diagnosis is confirmed by measurement of either intact glycoprotein hormones or their α and β subunits. Levels of the α subunit tend to be inappropriately elevated, compared with those of the intact hormone itself.11
HYPOPITUITARISM
Causes of Hypopituitarism
Lymphocytic Hypophysitis
Lymphocytic hypophysitis is an autoimmune disease often presenting in women during or after pregnancy. The clinical manifestations are secondary to hypopituitarism or adrenal insufficiency or are due to a pituitary mass effect. Serum prolactin is elevated in half of patients but may be decreased. Antipituitary antibodies are present in some patients, and other autoimmune endocrine disorders, including Hashimoto’s thyroiditis and Addison’s disease, have been seen by others.12
Recent experience from Germany suggests that one should be cautiously optimistic about high-dose steroid therapy. MRI findings improved in 88% of patients treated with high-dose steroids, but clinical normalization was quite variable, with none achieving complete recovery.13
Surgery for mass effect in lymphocytic hypophysitis can lead to rapid relief of neurologic symptoms, but endocrinologic improvement was seldom reported. Indications for surgery are the presence of gross chiasma compression, ineffectiveness of corticosteroid therapy, and the impossibility of establishing the diagnosis of lymphocytic hypophysitis with sufficient certainty by conservative evaluation.14
Sheehan’s Syndrome
Sheehan’s syndrome is the result of ischemic infarction of the normal pituitary gland, leading to hypopituitarism secondary to postpartum hemorrhage and hypotension.15 Patients have a history of failure to lactate postpartum, failure to resume menses, cold intolerance, or fatigue. Some women may have an acute crisis mimicking apoplexy within 30 days postpartum. There is often subclinical central diabetes insipidus.16
ADRENAL GLAND DISORDERS
Anatomy and Physiology
The zonae reticularis and fasciculata are under the control of corticotropin released by the pituitary gland in response to hypothalamic CRH. CRH in turn is regulated by cortisol-negative feedback, stress, and a circadian rhythm. Besides increasing the synthesis of cortisol, corticotropin is trophic for the adrenal gland so that a lack of corticotropin results in atrophy of the zonae fasciculata and reticularis. Although corticotropin has some effect on aldosterone production, the zona glomerulosa is predominantly under the control of renin. Understanding the anatomy and physiology of the adrenal gland is crucial to understanding its hypofunction and hyperfunction.17
Adrenal Insufficiency
Etiology
Clinical adrenal insufficiency results from hypofunction of the adrenal cortex. This may be due to destruction of the adrenal gland itself, referred to as Addison’s disease or primary adrenal insufficiency. Alternatively, it may be due to a lack of either corticotropin (i.e., secondary adrenal insufficiency) or CRH.18
The most common cause of Addison’s disease in adults (80%) is autoimmune destruction of the adrenal gland. This is often seen in association with other autoimmune diseases, including Hashimoto’s thyroiditis, Graves’ disease, or type 1 diabetes mellitus. Adrenal insufficiency in this setting is known as type II autoimmune polyglandular syndrome. Type I autoimmune polyglandular syndrome, more commonly seen in children, consists of Addison’s disease, hypoparathyroidism, and mucocutaneous candidiasis.
Other causes associated with adrenal insufficiency are listed in Table 22-6. Currently, acquired immunodeficiency syndrome is the most common cause of infectious adrenal destruction, and the antiphospholipid syndrome (lupus anticoagulant) is increasingly being recognized as a cause of adrenal hemorrhage.
Table 22-6 Other Causes of Primary Adrenal Insufficiency in Adults
| Hemorrhage/infarction |
| Anticoagulants/coagulopathy |
| Sepsis |
| Thrombosis |
| Metastatic cancer: breast, lung, gastrointestinal, renal |
| Infiltrative disorders: amyloidosis, sarcoidosis, hemochromatosis |
| Central nervous system demyelination |
Secondary adrenal insufficiency is a result of adrenal gland atrophy from corticotropin deficiency. This most often results from pituitary corticotroph atrophy owing to previous exogenous glucocorticoid use,19 hypopituitarism, or isolated corticotropin deficiency (usually postpartum).
Clinical Presentation
The underlying etiology of adrenal insufficiency determines the clinical presentation (Table 22-7). Under the regulation of corticotropin, cortisol and adrenal androgens are lost in both primary and secondary adrenal insufficiency. Aldosterone production, predominantly regulated by renin, remains intact in secondary adrenal insufficiency. Therefore, hyperkalemia and profound dehydration with orthostatic hypotension are seen in primary adrenal insufficiency only. Likewise, hyperpigmentation of the skin or mucous membranes (secondary to increased corticotropin) is seen in primary adrenal insufficiency only. The absence of hyperkalemia or hyperpigmentation does not exclude adrenal insufficiency. In addition to hyponatremia and hyperkalemia, laboratory abnormalities in adrenal insufficiency may include hypoglycemia (usually chronic), hypercalcemia, eosinophilia, and lymphocytosis.
Table 22-7 Adrenal Insufficiency Signs and Symptoms
| Primary | Secondary | |
|---|---|---|
| Yes | Yes | |
| Yes | Yes | |
| Yes | No | |
| Yes | No |
Diagnosis
In secondary adrenal insufficiency, however, the corticotropin stimulation test is not always abnormal. Adequate corticotropin may be present to prevent adrenal gland atrophy, thereby resulting in a response to the supraphysiologic dose of corticotropin used in the test. However, the HPA axis may not be able to respond to stress. In patients with suspected secondary adrenal insufficiency and a normal corticotropin stimulation test, CRH is now available to assess corticotropin response. In addition, the insulin tolerance test or the metyrapone test evaluate the integrity of the HPA axis by its response to hypoglycemia or inhibited cortisol synthesis, respectively. Although not widely used, some investigators find that a 1μg corticotropin stimulation test may be more sensitive at detecting mild adrenal insufficiency.20
Treatment
Cortisol 20mg in the morning and 10mg in the evening, or prednisone, 5 to 7.5mg daily, provides dramatic relief of symptoms. However, to prevent Cushing’s syndrome, the smallest dose needed to control the patient’s symptoms should be used. For a minor illness, the glucocorticoid dose should be doubled for as short a time as needed. For a major stress, parenteral hydrocortisone, 200 to 400mg daily, is given initially and then rapidly tapered. Aldosterone replacement is required in primary adrenal insufficiency only and is given as fludrocortisone acetate (Florinef Acetate), 0.05 to 0.2mg daily. The dose is adjusted according to the blood pressure and potassium level. Renin levels may be required to assess plasma volume. Adrenal androgens are not replaced.21
In undiagnosed patients with suspected adrenal crisis, dexamethasone, 2 to 4 mg intravenously or intramuscularly, should be given along with saline and glucose. Dexamethasone does not interfere with the cortisol assay. The corticotropin stimulation test should then be done as soon as possible.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


