Chapter 74 Management of ascites in cirrhosis and portal hypertension
Historic Overview
One of the most visible manifestations of liver disease is that of ascites, broadly defined as the pathologic accumulation of fluid within the peritoneal cavity (Runyon, 2009). The almost reflexive association of ascites with liver disease notwithstanding, ascitic fluid volume and composition demonstrate a variability that reflects the range of disease processes, from benign to sinister, from whence it originates.
Descriptions of ascites exist in both human history and prehistory. Indian medical treatises from 1500 bce, Mayan figurines with protuberant abdomens and everted umbilici, and the writings of Hippocrates all testify to the experience of varied cultures with this problem. Even the term ascites—derived from the Greek askos, a bag made of leather or sheepskin used to contain liquids—reflects its ancient origins (Reuben, 2004).
High-volume ascites is readily evident on physical examination; however, smaller volumes may evade detection, particularly in the obese. Ultrasound is the best imaging modality for detection of ascites. It is inexpensive, avoids ionizing radiation, and has the added benefit of providing information on liver architecture and portal vein patency (see Chapter 13).
All clinicians should maintain a low threshold for performing paracentesis. The procedure is necessary in the diagnostic evaluation of newly developed ascites, or when there is a change in the clinical condition in an individual with cirrhosis and ascites. A percutaneous approach that avoids the epigastric vessels offers an avascular plane that is safe even in coagulopathic and thrombocytopenic patients (Wong et al, 2008).
Portal Hypertension and Mechanisms of Ascites Formation
Ascites is the most common complication of portal hypertension arising from cirrhosis, occurring at an annual incidence of 1% (see Chapter 70A, Chapter 70B ; Ginès et al, 1987). Its development heralds a significant change in clinical condition, with a median survival of 50% over 2 years (D’Amico et al, 1986). Portal hypertension can arise from cirrhotic and noncirrhotic causes, although as a manifestation of portal hypertension, ascites is most common in disorders that increase pressures within the hepatic sinusoids, either from sinusoidal processes (cirrhosis) or postsinusoidal processes (heart failure, hepatic venous obstruction). By comparison, ascites is uncommon in presinusoidal portal hypertension.
The normal liver architecture consists of sinusoids that convey blood from the portal vein to the central vein. The sinusoids are separated from cords of hepatocytes by a fenestrated endothelium through which oxygen, cells, and plasma components are permitted to diffuse. The principal collagen-producing cells of the liver, the stellate cells, reside within the space of Disse, and activation of these cells leads to collagen deposition (Friedman et al, 1992). Other cells that contribute to the extracellular matrix within the liver are bone marrow–derived myofibroblasts and fibroblasts derived from epithelial-mesenchymal transition (Forbes et al, 2004; Kalluri & Neilson, 2003).
In cirrhosis, extracellular basement membrane deposition of collagen fibers within the space of Disse results in capillarization of the hepatic sinusoids (Huet et al, 1982). The resulting architectural changes cause a static increase in pressure within the splanchnic circulation (see Chapter 6).
The endothelial cells of the liver also play an important role in controlling dynamic changes to the hepatic microcirculation through the elaboration of nitric oxide (Mittal et al, 1994; Shah et al, 1997) and expression of endothelin-1 receptors (Bauer et al, 2000). Decreased endothelial nitric oxide synthase (eNOS) activity (Gupta et al, 1998; Shah et al, 1999), increased eNOS inactivator activity (Liu et al, 2005), and endothelin-1 overexpression in cirrhosis (Pinzani et al, 1996) have all been proposed as mechanisms that promote endothelial cell dysfunction in cirrhosis (see Chapter 6).
In the splanchnic circulation outside of the hepatic environment, different but equally important changes occur that contribute to portal hypertension. The most prominent of these is splanchnic arterial vasodilation. In experimental models of cirrhosis, vasodilation is mediated by NO-dependent (Sieber & Groszmann, 1992; Sieber et al, 1993) and NO-independent processes. The NO-independent processes include those related to endogenous vasodilatory cannabanoids (Garcia et al, 2001) and overexpression of vascular endothelial growth factor (VEGF) and VEGF receptor-2 to promote splanchnic angiogenesis, thereby augmenting blood flow in the splanchnic circulation (Fernandez et al, 2007). Vasodilation in the splanchnic circulation would decrease the effective arterial circulation if not for compensatory increases in cardiac output (Iwakiri & Groszmann, 2006).
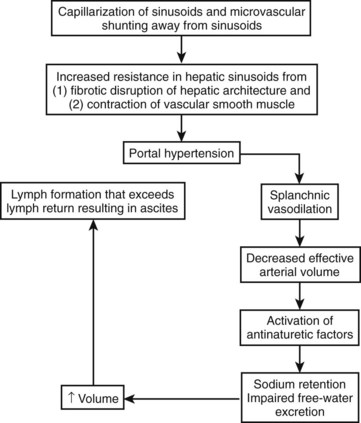
With progression of portal hypertension, other compensatory mechanisms are activated to maintain the arterial circulation in the face of even greater increases in vasodilation and declines in cardiac output. These mechanisms include activation of the renin-angiotensin system and sympathetic nervous system to stimulate sodium retention by the kidneys (Arroyo et al, 1983). The nonosmotic release of arginine vasopressors is an additional compensatory mechanism to increase the effective arterial volume, even at the expense of tonicity; this is reflected in the development of hyponatremia (Arroyo et al, 1994). The cumulative effect of increased hydrostatic pressure in the hepatic microcirculation in the setting of increased splanchnic volume is that of hepatic lymph formation in excess of its removal; the resulting excess fluid weeps into the peritoneal cavity and is recognized as ascites (Fig. 74.1).
Characteristics and Evaluation of Ascites
Ascitic fluid in cirrhotics is transparent but will generally take on a yellow or amber color. The fluid generally has a low leukocyte (<100 µL/mm) and red blood cell content. The fluid protein is typically less than 2.5 mg/dL, and the protein content varies inversely with the severity of the portal hypertension (Hoefs, 1983). Measurement of the serum albumin ascites gradient (SAAG) is both a highly accurate and clinically facile technique for assessing the origins of ascites. The SAAG is calculated by subtracting the concentration of albumin in the ascites from that in the plasma (Pare et al, 1983). With an approximately 97% accuracy, a gradient greater than 1.1 g/dL indicates the presence of portal or sinusoidal hypertension (Runyon et al, 1992).
Opacification of ascitic fluid can arise from a number of disparate processes. Bloody ascites (hematocrit >0.5) can be seen in traumatic paracentesis or in the spontaneous rupture of hepatocellular cancer. Chylous ascites is milky in appearance from increased concentration of chylomicron-rich triglyceride (Aalami et al, 2000). It arises from processes that disrupt lymphatic flow, most commonly lymphangiectasia and lymphoma, but it can also occur with abdominal trauma and surgical disruption of the cysterna chyli. Cirrhotic ascites can also take on a chylous appearance, owing to rupture of abdominal lymphatics from portal hypertension (Rector, 1984). In these cases, known as pseudochylous ascites, the triglyceride concentration is generally less than the threshold value of 110 mg/dL found in pure cases of chylous ascites.
Both malignancy and tuberculosis peritonitis can result in ascites, and in both circumstances the SAAG is less than 1.1 g/dL; however, confusion may arise when liver disease coexists, as in the case of tuberculosis and alcohol-related liver disease (Aguado et al, 1990; Shakil et al, 1996). The diagnosis of malignant ascites is established by the finding of cancer cells within the peritoneal cavity. This can be done by conventional cytology with a diagnostic sensitivity of 40% to 60% (Siddiqui et al, 1992). The accuracy of cytology can be improved when combined with immunohistologic staining (Aslam & Marino, 2001).
The peritoneum is a common site of involvement of tuberculosis (TB), and in the United States, the peritoneum is the sixth most common extrapulmonary site. Peritoneal cell counts typically vary between 500 and 1500 cells/mm3 with a lymphocyte predominance in 68% (Sanai & Bzeizi, 2005), although the absence of a lymphocyte predominance does not exclude TB, particularly in patients with underlying renal failure, in whom the cells are mostly neutrophils (Lui et al, 2001). Mycobacterial culture of the fluid has a diagnostic sensitivity of 34% and requires several weeks of intubation. Measurement of adenosine deaminase activity in the peritoneal fluid has been proposed as another diagnostic test with high sensitivity and specificity, although the positive predictive value has been reported to be low in the setting of concomitant cirrhosis (Hillebrand et al, 1996). Of all diagnostic strategies, laparoscopy with peritoneal biopsy affords the highest sensitivity and specificity and permits exclusion of other granulomatous and nongranulomatous processes that can produce a low-SAAG ascites. The ascites concentration of lactate dehydrogenase (LDH) tends to be higher than that of serum LDH in malignant ascites and less than half that of serum in tuberculous peritonitis.
Management
Dietary Sodium Restriction
Avid renal sodium retention is the initial response to splanchnic arterial vasodilation; thus initial treatment strategies involve tipping the balance in favor of a net loss of sodium; this is most simply accomplished with dietary sodium restriction. The estimated mean sodium consumption among nonhypertensive adults in the United States is 3600 mg/day (Ajani et al, 2005). Those with ascites are commonly advised to restrict dietary salt intake to 1.5 to 2 g per day (67 to 87 mmoL/day), the lower value of which is considered adequate for daily needs. In patients with mild degrees of ascites, sodium restriction may be singularly effective. This group usually has baseline rates of sodium excretion of at least 40 mEq/L per day and normal plasma sodium concentrations (Arroyo et al, 1981). Although seemingly a simple intervention, success with dietary salt restriction requires counseling and vigilance. Most of the sodium present is added during food processing, and the patient’s actual consumption may not be apparent to them unless food labeling is read and understood (Cook, 2008).
Medical Management
Diuretics
Most patients will at some point require diuretics. Between 500 and 750 mL per day of ascites can be mobilized without intravascular depletion, and greater amounts of ascitic fluid losses may be tolerable in the presence of edema, which tends to act as a buffer (Pockros & Reynolds, 1986). The avoidance of rapid fluid loss is critical because precipitous and excessive volume contraction can give rise to hepatorenal syndrome.
Aldosterone Antagonists
The aldosterone antagonists spironolactone and amiloride (Table 74.1) can be used as either monotherapy or in combination with loop diuretics. These agents prevent sodium reabsorption in the distal tubule and cortical collecting duct. Although aldosterone antagonists are weak natriuretics, they are effective in patients with cirrhosis (Perez-Ayuso et al, 1983).
Table 74.1 Diuretics and Dosages Commonly Used in the Management of Ascites
| Medication | Dose | Comments |
|---|---|---|
| Aldosterone Antagnosists | ||
| Spironolactone | 50-400 mg daily | |
| Amiloride | 5-10 mg daily | Suitable substitute when spironolactone use is associated with painful gynecomastia |
| Loop Diuretics | ||
| Furosemide | 20-160 mg daily | Avoid intravenous use |
Urine sodium excretion and plasma aldosterone concentration are hyperbolically and inversely related in patients with cirrhosis, and a greater sensitivity to the dose-response curve is observed in those with ascites (Bernardi et al, 1983). One explanation for the effectiveness of these agents in cirrhotic ascites may be that they target the functional hyperaldosteronism that would otherwise permit sodium reabsorption in the cortical collecting tube of fluid filtered in the loop of Henle. In addition, unlike other diuretics that require access to the tubular lumen, spironolactone enters the principal cell of the collecting duct from the plasma compartment and thereby circumvents decreases in renal blood flow and hypoalbuminemia, commonly encountered in patients with cirrhosis, that might otherwise impair its activity (Horisberger & Giebisch, 1987).
Loop Diuretics
As monotherapy for ascites, loop diuretics are often unsuccessful. The reasons for this are unclear but may relate to a decreased rate of drug entry into the tubular lumen or to a compensatory increase in distal tubular sodium resorption mediated by aldosterone. By comparison, the combination of loop diuretics and aldosterone antagonists is the most commonly used combination for moderate to severe ascites and can achieve reductions in ascites beyond that of aldosterone antagonists alone. The most commonly used loop diuretic is furosemide (see Table 74.1), beginning at doses of 20 to 40 mg daily. The dose is doubled in a coordinated fashion with increases in spironolactone, until therapeutic effect is achieved. Like spironolactone, a fourfold doubling of the dose (160 mg daily) is considered a maximal dose. An added benefit of loop diuretics is that they counteract the hyperkalemia associated with aldosterone antagonists, and they may also benefit those who develop dependent edema in addition to ascites.
Albumin
Hypoalbuminemia is a frequent finding in patients with cirrhosis, and it may influence the development of edema and ascites through alterations in plasma oncotic pressure and the activity of loop diuretics in the lumen of the collecting tubule. Limited evidence supports a role for albumin administration as an adjunct to diuretic therapy in ascites that is difficult to control (Gentilini et al, 1999). However, this strategy may not be broadly applicable with the diuretics currently available or when there is recourse to other treatments (Blendis & Wong, 1999). Intravenous albumin administration does have a secure role in the prevention of renal dysfunction, which occurs in the one third of patients who develop spontaneous bacterial peritonitis (Sort et al, 1999). In addition, vasoconstrictor agents used in the treatment of hepatorenal syndrome are more effective when coadministered with albumin than when administered with saline or other crystalloids (Ortega et al, 2002). Thus albumin appears to achieve a successful volume expansion in patients with cirrhosis, greater than that that of crystalloid, which is translatable into improvement in solid clinical end points.
Arginine Vasopression Receptor Antagonists
The nonosmotic release of antidiuretic hormone (ADH) is commonly observed in patients with cirrhosis as a compensatory mechanism for the decrease in effective arterial volume that occurs with splanchnic vasodilation. ADH exerts its effects through arginine vasopressin receptor-2 (AVPR2), which is predominantly expressed in the distal convoluted tubule and collecting ducts of the kidney (van den Ouweland et al, 1992). Satavaptan is a selective AVPR2 receptor antagonist that has been tested in clinical studies of both the syndrome of inappropriate ADH release (Soupart et al, 2006) and hyponatremia occurring with cirrhosis (Ginès et al, 2008). The study in patients with cirrhosis was a multicenter, double-blind, randomized controlled study of 110 subjects with ascites and hyponatremia. Treatment with satavaptan and spironolactone was associated with improvement in ascites volume concomitant with improvements in hyponatremia more often than the placebo. The use of satavaptan and other aquaretic agents represents a promising area for pharmacologic management of ascites if these early successes are confirmed in other clinical trials.
Refractory Ascites
Ascites that persists despite dietary sodium restriction and high-dose diuretics (spironolactone 400 mg/day and furosemide 160 mg/day) is referred to as refractory ascites (Arroyo et al, 1996). Diuretic-intolerant ascites describes diuretic failure because of intolerant side effects—such as azotemia, hyponatremia, and encephalopathy—that prevent the attainment of a dose sufficient to effect an adequate ascites loss. The clinical significance of refractory ascites should not be overlooked; the survival curves of individuals with refractory ascites approximate those with type II hepatorenal syndrome (Ginès et al, 2004).
Before labeling a patient as having refractory ascites, it is important to exclude excessive sodium intake and other medications that may influence the diuretic response. In particular, nonsteroidal antiinflammatory agents, including aspirin (Planas et al, 1983), may both decrease the response of diuretics and contribute to azotemia.
Treatment options for refractory ascites includes therapeutic paracentesis, transjugular intrahepatic portosystemic shunting (TIPS) (see Chapter 76E), peritoneal shunting, and liver transplantation (see Chapter 97A).
Paracentesis
High-volume paracentesis, also known as therapeutic paracentesis, was a technique known to and practiced by physicians as far back as ancient Greece. It was also the most effective treatment in practice before the development of modern diuretics, after which the practice fell out of favor until it was reintroduced in 1987 as safe and effective (Ginès et al, 1987).
Related posts:
 Preoperative and postoperative nutrition in hepatobiliary surgery
Preoperative and postoperative nutrition in hepatobiliary surgery
 Portal hypertension in children
Portal hypertension in children
 Liver transplantation for cholangiocarcinoma and other neoplastic diseases
Liver transplantation for cholangiocarcinoma and other neoplastic diseases
 Distal splenorenal shunt
Distal splenorenal shunt
 Magnetic resonance imaging of the liver, biliary tract, and pancreas
Magnetic resonance imaging of the liver, biliary tract, and pancreas
 Interventional endoscopy: Technical aspects
Interventional endoscopy: Technical aspects
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


