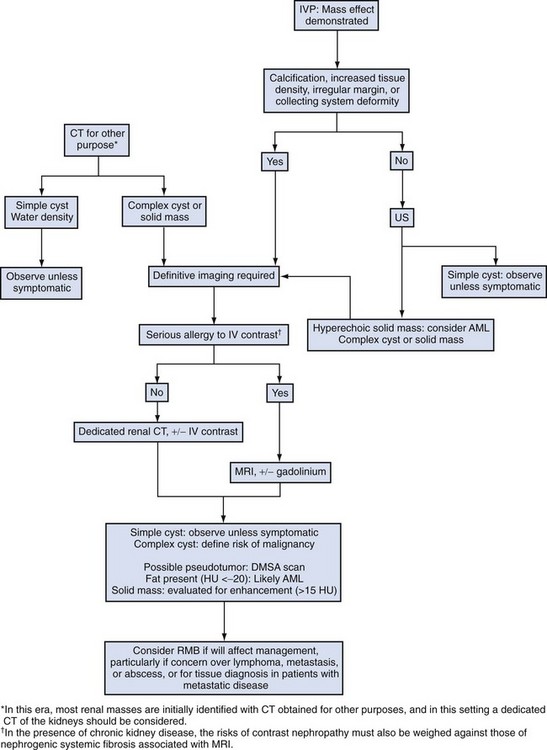
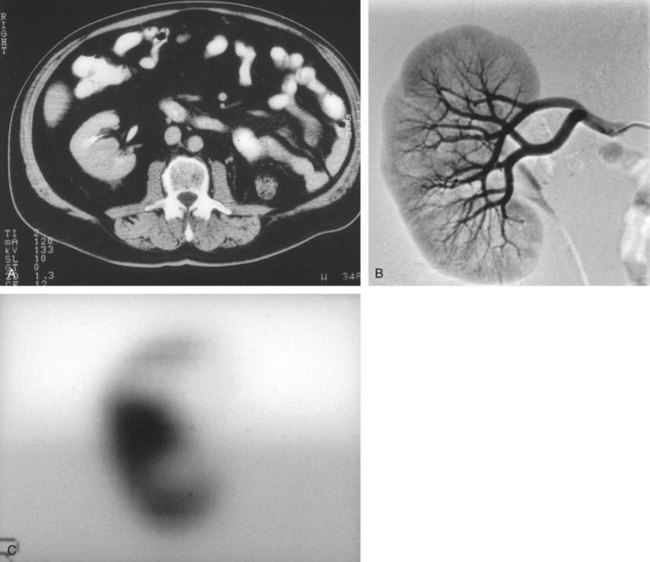
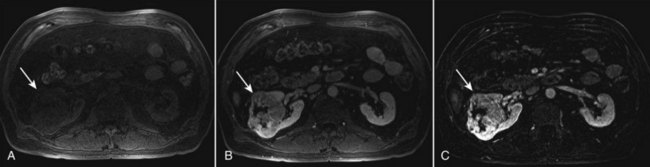
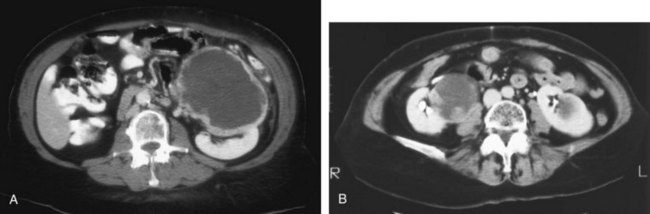
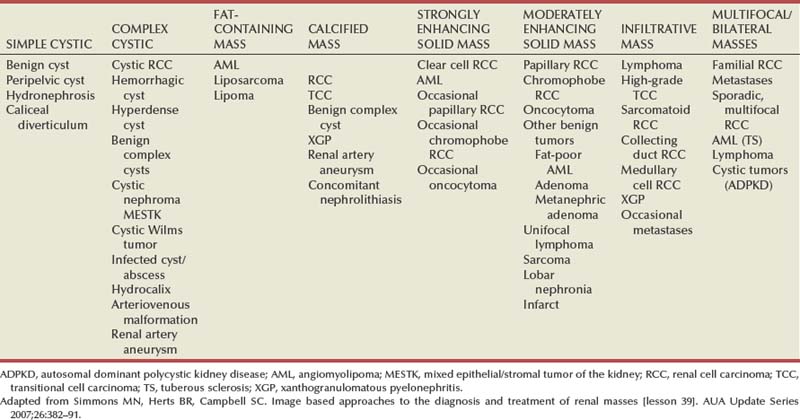
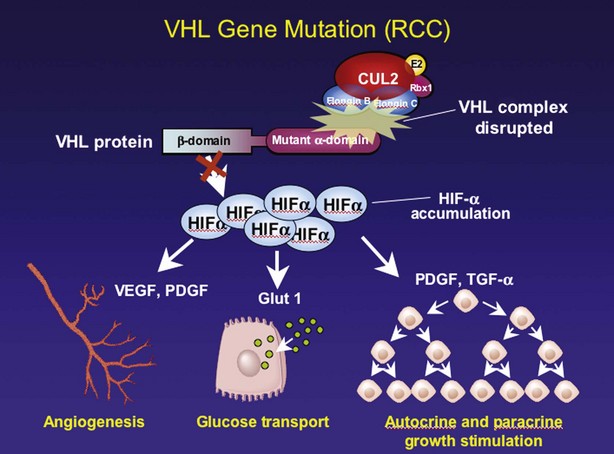
Steven C. Campbell, MD, PhD, Brian R. Lane, MD, PhD The introduction of nephrectomy and other subsequent surgical interventions for renal diseases provided the clinical information and histopathologic insight that form the basis of current concepts regarding renal tumors. The first documented nephrectomy was accomplished in 1861 by Wolcott, who operated with the mistaken assumption that the tumor mass was a hepatoma. In 1867, Spiegelberg removed a kidney incidentally in the course of excising an echinococcus cyst. The first planned nephrectomy was performed by Simon in 1869 for persistent ureteral fistula, and this patient survived with cure of the fistula. One year later (1870), the first planned nephrectomy in the United States was successfully accomplished by Gilmore in Mobile, Alabama, for treatment of atrophic pyelonephritis and persistent urinary tract infection (Glenn, 1980; Herr, 2008). Harris (1882) subsequently reported on 100 surgical extirpations of the kidney, a sufficient number to permit analysis of clinical, surgical, and pathologic features of renal disorders that require surgery. With surgical intervention, tissue became available to pathologists for histologic interpretation. Unfortunately, such interpretation was not always accurate, and there were often serious professional differences of opinion. According to Carson (1928), the first accurate gross description of kidney tumors dates to 1826, with Konig’s observations. In 1855, Robin examined solid tumors apparently arising in the kidney and concluded that renal carcinoma arose from renal tubular epithelium. This interpretation was confirmed by Waldeyer in 1867. Unfortunately, theoretical and practical considerations of renal tumors were confused by Grawitz (1883), who contended that such apparent renal tumors arose from adrenal rests within the kidney. He introduced the terminology struma lipomatodes aberrata renis as descriptive nomenclature for the tumors of clear cells that he believed were derived from the adrenal glands. He based his conclusions not only on the fatty content of the tumors, analogous to that seen in the adrenal glands, but also on the location of the tumors beneath the renal capsule, the approximation to the adrenal glands, the lack of similarity of the cells to uriniferous tubules, and the demonstration of amyloid similar to that seen with adrenal degeneration. This histogenetic concept was adopted by subsequent investigators, and pathologists of the era readily embraced the idea that renal tumors truly arose from the adrenal glands. In 1894, Lubarch endorsed the idea of a suprarenal origin of renal tumors, and the term hypernephroid tumors, indicating origin above the kidneys, was advocated by Birch-Hirschfeld (Birch-Hirschfeld and Doederlein, 1894). This semantic and conceptual mistake led to the introduction of the term hypernephroma, which predominated in the literature describing parenchymal tumors of primary renal origin. Some clarification of the histopathology of renal tumors was derived from the work of Albarran and Imbert (1903), and the four-volume contribution of Wolff (1883), written between 1883 and 1928, added further historical significance to the understanding of renal tumors (Glenn, 1980). The modern era has brought an appreciation that renal cell carcinoma (RCC) includes a number of distinct subtypes derived from the various parts of the nephron, each with a unique genetic basis and tumor biology (Linehan et al, 2003, Cohen and McGovern, 2005; Novick, 2007; Rini et al, 2009). Other major advances in the past several decades have included the introduction of radical nephrectomy followed by a trend toward nephron-sparing surgery and, more recently, a variety of minimally invasive approaches (Robson, 1963; Uzzo and Novick, 2001; Abreu and Gill, 2003; Herr, 2005, 2008; Novick, 2007). One common theme that has persisted is that RCC remains primarily a surgical disease—it is still considered the paradigm of the chemorefractory tumor; and although immune-based and targeted molecular approaches have shown promise, overall response rates remain low (Négrier et al, 2002; Milowsky and Nanus, 2003; Escudier et al, 2007; Motzer et al, 2007; Rini et al, 2009). Unfortunately, the incidence of RCC is gradually increasing; and despite a trend toward earlier detection, mortality rates remain high. Renal masses can be malignant, benign, or inflammatory as classified by Barbaric (1994) (Table 49–1), or they can be classified based on radiographic appearance (simple cystic, complex cystic, fatty tumors, and others) (Table 49–2). These classification schemes have been updated taking into account current knowledge about the distinct subtypes of RCC and recent advances in our understanding of the various benign and malignant tumors of the kidney (Eble et al, 2004; Cohen and McGovern, 2005; Rini et al, 2009). This approach is practical and should assist in the differential diagnosis of renal masses. Malignant renal tumors include RCC, urothelial-based lesions (addressed elsewhere), sarcomas, embryonic or pediatric tumors, and lymphomas and metastases. Benign renal tumors are diverse and present unique diagnostic challenges (see Chapter 51). Inflammatory and vascular lesions must also be considered in the differential diagnosis. Table 49–1 Renal Masses Classified by Pathologic Features Several radiographic modalities are currently available for detection and evaluation of renal masses, each with relative strengths and limitations (Davidson et al, 1997; Zagoria, 2000; Israel and Bosniak, 2003a; Zhang et al, 2007a; Herts, 2009). A systematic approach is necessary to ensure diligent evaluation of suspected renal masses, given the large differential diagnosis and considerable overlap between benign and malignant renal lesions (Fig. 49–1; see Table 49–2) (Ananthakrishnan et al, 2007; Simmons et al, 2007). Although intravenous pyelography was often the first test that indicated a renal mass in the past, it is now only occasionally used for the evaluation of hematuria. The lack of sensitivity and specificity of intravenous pyelography for the detection of parenchyma tumors is well documented. In particular, intravenous pyelography may miss small anterior or posterior lesions that do not distort the collecting system or the contour of the kidney. Features suggestive of malignancy on intravenous pyelography include calcification within the mass, increased tissue density, irregularity of the margin, and distortion of the collecting system (Zagoria, 2000). When a renal mass is identified by intravenous pyelography, unless the mass has features suggestive of malignancy, ultrasonography should be the next study performed because it is noninvasive, accurate, and relatively inexpensive (Davidson et al, 1997; Paspulati and Bhatt, 2006). Ultrasonography is reliable for differentiation of solid tissue from fluid and can establish the diagnosis of a simple renal cyst. Strict ultrasonographic criteria for simple cysts have been defined and include a smooth cyst wall, a round or oval shape without internal echoes, and through-transmission with strong acoustic shadows posteriorly. If these criteria are met, observation is sufficient in an asymptomatic patient. In evaluating complicated renal cysts, important ultrasonographic features include thickness and contour of the cyst wall, number and thickness of any septa, presence of any calcifications, density of the renal cyst fluid, and presence of solid components. ultrasonography is helpful in suggesting the fat content of an angiomyolipoma (AML) by its characteristic increased echogenicity (Nelson and Sanda, 2002). A renal mass that is not clearly a simple cyst by strict ultrasound criteria should be evaluated further with computed tomography (CT). A dedicated (thin-slice) renal CT scan remains the single most important radiographic test for delineating the nature of a renal mass. CT, with and without the administration of contrast material, is necessary to take full advantage of the contrast enhancement characteristics of highly vascular renal parenchymal tumors (Davidson et al, 1997; Zagoria, 2000, Prasad et al, 2008; Ng et al, 2008; Zhang et al, 2007a). In general, any renal mass that enhances with intravenous administration of contrast material on CT by more than 15 Hounsfield units (HU) should be considered an RCC until proved otherwise (Fig. 49–2) (Hartman et al, 2004). Solid masses that also have substantial areas of negative CT attenuation numbers (below −20 HU) indicative of fat are diagnostic of AMLs (Nelson and Sanda, 2002). In 10% to 20% of solid renal masses CT findings are indeterminate, and additional testing or surgical exploration is needed to establish a definitive diagnosis. On occasion, CT demonstrates an enhancing renal segment that is isodense with the remainder of the kidney, suggestive of a renal pseudotumor. Renal pseudotumors may be due to a hypertrophied column of Bertin, renal dysmorphism, or an unusually shaped kidney (Bhatt et al, 2007). In this situation, the diagnosis of a pseudotumor can be confirmed by isotope renography with technetium-labeled dimercaptosuccinic acid or glucoheptonate (Fig. 49–3). These isotope studies demonstrate an area of increased density if the mass is a pseudotumor and an area of decreased density if the mass is a cyst or solid tumor (Israel and Bosniak, 2003a). (Courtesy of Dr. Terrence Demos, Maywood, IL.) Magnetic resonance imaging (MRI) is the alternate standard imaging modality for the characterization of a renal mass (Pretorius et al, 2000; Zhang et al, 2004; Bassignani, 2006). A basic consideration in the evaluation of a renal mass is that for such a mass to be considered malignant it must enhance with the intravenous administration of contrast material. Such enhancement can now be determined equally well by magnetic resonance angiography with intravenous gadolinium-labeled diethylenetriaminepentaacetic acid, although the assessment is qualitative rather than quantitative. On T1-weighted scans before and after administration of gadolinium, enhancement (vascularity) of the mass is detected (Fig. 49–4). This technique is most helpful in patients for whom iodinated contrast medium is contraindicated because of severe allergy. One concern with MRI with gadolinium is the uncommon but potentially serious complication of nephrogenic systemic fibrosis (NSF), which is more common in patients with renal insufficiency (Bach and Zhang, 2008). Current recommendations are to avoid MRI, particularly serial studies, in this population whenever possible, and to dialyze patients after the study if severe chronic kidney disease (CKD) is present. MRI was previously the imaging procedure of choice in patients with CKD and a renal mass. Now many radiologists prefer CT with intravenous contrast and careful periprocedural hydration, but decision making must be individualized. Contrast-enhanced ultrasonography using microbubbles has also shown promise for the safe characterization and assessment of enhancement of renal masses and may play an important role in patients with CKD in the future (Simmons et al, 2007). Renal mass biopsy is now being revisited for the evaluation of renal masses (Wunderlich et al, 2005; Lane et al, 2007b; Maturen et al, 2007; Volpe et al, 2007; Lebret et al, 2007; Strope and Wolf, 2008; Wang et al, 2009). Historically the false-negative rate of renal mass biopsy was thought to be 18%, too high to justify routine use. However, most of these “false negatives” were in reality instances in which the mass could not be adequately targeted or the material obtained was insufficient for the pathologist to make a definitive determination. Review of this literature has shown that, although about 15% of renal mass biopsy specimens are nondiagnostic, the real false-negative rate was only 4% in studies prior to 2001 and less than 1% since then (Lane et al, 2007b). Overall accuracy is greater than 80%. Assessment of tumor grade and histologic type, which reflects tumor aggressiveness, is also accurate in the majority of cases (Barocas et al, 2006; Schmidbauer et al, 2008). The risks of clinically significant perinephric bleeding and pneumothorax also appear to be low (<1%), and needle tract seeding is exceedingly rare when centrally located, infiltrative renal masses are excluded. Poorly differentiated transitional cell carcinoma is much higher risk for needle tract seeding than RCC. Given the great heterogeneity in the tumor biology of enhancing clinical T1 renal masses, with 20% benign, 60% indolent RCC, and 20% potentially aggressive RCC, renal mass biopsy is now being considered more frequently, particularly in patients who are potential candidates for a wide variety of treatment options ranging from observation to surgical excision. Younger, healthy patients who are unwilling to accept the uncertainty associated with renal mass biopsy are still typically managed primarily based on radiographic and clinical considerations. More traditional indications for renal mass biopsy include suspicion of renal abscess or when RCC must be differentiated from metastatic malignant disease or renal lymphoma (Herts and Remer, 2000; Anderson et al, 2006; Vasudevan et al, 2006; Beland et al, 2007; Kummerlin et al, 2008a; Somani et al, 2007; Shannon et al, 2008; Volpe et al, 2008). The differentiation between a benign renal cyst and a cystic RCC remains one of the more common and difficult problems in renal imaging (Balci et al, 1999; Harada et al, 2002; Kausik et al, 2002; Israel and Bosniak, 2005; Warren and McFarlane, 2005). When a complex renal cyst is identified, determination of its benign or malignant nature is based on evaluation of the wall of the lesion; its thickness and contour; the number, contour, and thickness of any septa; the amount, character, and location of any calcifications; the density of fluid in the lesion; the margination of the lesion; and the presence of solid components. Bosniak developed a useful classification scheme primarily based on CT imaging criteria that divides renal cystic lesions into categories that are distinct from one another in terms of the likelihood of malignancy (Bosniak, 1997; Israel and Bosniak, 2005). Category I lesions are uncomplicated, simple, benign cysts of the kidney that are straightforward to diagnose on ultrasonography, CT, or MRI. These are by far the most common renal cystic lesions, and in the absence of associated symptoms, no treatment is necessary. Category II lesions are minimally complex cysts that are generally benign but have some radiologic findings that cause concern (Fig. 49–5). These lesions include septated cysts, cysts with calcium in the wall or septum, infected cysts, and hyperdense (high-density) cysts (Bosniak, 1997; Israel and Bosniak, 2005). Hyperdense cysts are benign lesions that contain old, degenerated, or clotted blood; therefore, the CT attenuation of their contents is increased (>20 HU). Classic hyperdense renal cysts are small (<3 cm), round, and sharply marginated and do not enhance after the administration of contrast material (Fig. 49–6). This category has now been subdivided to differentiate category II lesions that do not require surveillance from category IIF lesions that mandate surveillance. The nuances involved in this classification are highlighted in Table 49–3. High-quality imaging, preferably CT, and considerable radiologic expertise are required to optimize the characterization of complex renal cystic lesions. The risk of malignancy for category IIF renal cysts is 5% to 10%, and these lesions should be observed with periodic renal imaging (Kausik, 2002; Israel and Bosniak, 2003b, 2005). (Courtesy of Dr. Terrence Demos, Maywood, IL.) (Courtesy of Dr. Terrence Demos, Maywood, IL.) Category III lesions are more complex renal cysts that cannot be confidently distinguished from malignant neoplasms (Kausik, 2002; Israel and Bosniak, 2005). The radiographic features include thickened irregular or smooth walls or septa in which measurable enhancement can be observed (Fig. 49–7). In the absence of a mitigating factor such as renal trauma or infection, surgical exploration is usually indicated in healthy patients. About 50% of these lesions are malignant; the remainder prove to be benign multiloculated, hemorrhagic, or densely calcified cysts (see Table 49–2). Fine-needle aspiration of complex cysts is rarely performed because of concern about sampling error and tumor cell spillage. (Courtesy of Dr. Terrence Demos, Maywood, IL.) Category IV lesions have large cystic components; irregular, shaggy margins; and, most important, solid enhancing portions that provide a definitive diagnosis of malignancy (Fig. 49–8) (Bosniak, 1997; Israel and Bosniak, 2003c, 2005). Category IV lesions are almost invariably cystic RCCs that, if localized, require surgical treatment. For radiographically detected solid renal masses, the differential diagnosis is extensive and includes conditions such as RCC, oncocytoma, AML, transitional cell carcinoma, metastatic tumor, abscess, infarct, vascular malformation, and renal pseudotumor (see Table 49–2). The diagnosis of most of these lesions can be established on the basis of the clinical presentation and the characteristic radiographic features (Table 49–4), occasionally combined with endourologic studies or needle biopsy of the mass (Simmons et al, 2007; Dyer et al, 2008). However, it is not possible to reliably distinguish RCC from benign renal neoplasms, including oncocytoma and fat-poor AML, with current diagnostic techniques. Ten to 20 percent of small, solid, CT-enhancing renal masses with features suggestive of RCC prove to be benign after surgical excision (Silver et al, 1997). Although oncocytoma is a benign tumor (see Chapter 51), it can be multifocal and is occasionally associated with RCC in the same or the opposite kidney (Licht, 1995; Dechet, 1999). RCC, which accounts for 2% to 3% of all adult malignant neoplasms, is the most lethal of the common urologic cancers. Traditionally, 30% to 40% of patients with RCC have died of their cancer, in contrast to the 20% mortality rates associated with prostate and bladder carcinomas (Landis et al, 1999; Pantuck et al, 2001b). Approximately 54,000 new diagnoses of RCC are made each year in the United States, and 13,000 patients die of disease (Russo et al, 2008, Carrizosa and Godley, 2009; Jemal et al, 2009). Overall, approximately 12 new cases are diagnosed per 100,000 population per year, with a male-to-female predominance of 3 : 2 (Landis et al, 1999; Wallen et al, 2007; DeCastro and McKiernon, 2008; Woldrich et al, 2008; Carrizosa and Godley, 2009). This is primarily a disease of the elderly patient, with typical presentation in the sixth and seventh decades of life (Pantuck et al, 2001b; Wallen et al, 2007). Incidence rates are 10% to 20% higher in African Americans for unknown reasons (Chow et al, 1999, Lipworth et al, 2006; Stafford et al, 2008). The majority of cases of RCC are believed to be sporadic; only 2% to 3% are familial (Lipworth et al, 2006). The incidence of RCC has increased since the 1970s by an average of 3% per year for whites and 4% per year for African-Americans, largely related to the more prevalent use of ultrasonography and CT for the evaluation of a variety of abdominal complaints (Chow et al, 1999; DeCastro and McKiernon, 2008; Kummerlin et al, 2008b). This trend has correlated with an increased proportion of incidentally discovered and localized tumors and with improved 5-year survival rates for patients with this stage of disease (Konnak and Grossman, 1985; Thompson and Peek, 1988; Kessler et al, 1994; Pantuck et al, 2001b; Parsons et al, 2001, Kane et al, 2008). However, other factors must also be at play because Chow and colleagues (1999) have documented a steadily increasing mortality rate from RCC per unit population since the 1980s, and this was observed in all ethnic and both sex groups. They reported that the incidence of advanced tumors per unit population has also increased; and although the proportion of advanced tumors has decreased, the mortality rate per unit population has still been negatively affected (Chow et al, 1999; Hock et al, 2002; Wallen et al, 2007; DeCastro and McKiernon, 2008). This suggests that a deleterious change in tumor biology may have occurred during the past several decades, perhaps related to tobacco use, dietary factors, or exposure to other carcinogens (Chow et al, 1999; Pantuck et al, 2001b; Hock et al, 2002; Parsons et al, 2002; Kane et al, 2008). RCC in childhood is uncommon, representing only 2.3% to 6.6% of all renal tumors in children (Castellanos et al, 1974; Chan et al, 1983; Freedman et al, 1996; Asanuma et al, 1999; Broecker, 2000). Mean age at presentation in children is 8 to 9 years, and the incidence is similar in boys and in girls. Although Wilms tumor is much more common in younger children, RCC is as common as Wilms tumor during the second decade of life. RCC in children and young adults is more likely to be symptomatic and to exhibit papillary histology, and a predilection for locally advanced, high-grade disease, and unfavorable histologic subtypes has also been reported (Freedman et al, 1996; Renshaw et al, 1999; Sánchez-Ortiz et al, 2004b; Cook et al, 2006; Estrada et al, 2005). TFE3 protein overexpression, which correlates with the presence of ASPL-TFE3 and PRCC-TFE3 gene translocation events involving the X and first chromosomes, is relatively common in children and young adults with RCC and is unique to this population (Heimann et al, 2001; Geller et al, 2008). The clinical significance of TFE3 protein overexpression is not well defined, although preliminary data suggest that these tumors may show differential sensitivity to certain chemotherapeutic agents (Argani et al, 2002; Heimann et al, 2001; Perot et al, 2003; Bruder et al, 2004). Most studies suggest that stage for stage, children and young adults with RCC may respond better to surgical therapy, and a number of long-term survivors have been reported after radical nephrectomy and lymphadenectomy for lymph node–positive disease (Freedman et al, 1996; Asanuma et al, 1999; Abou El Fettouh et al, 2002; Geller et al, 2008; Sánchez-Ortiz et al, 2004b; Geller et al, 2008). An aggressive surgical approach with formal lymphadenectomy has thus been recommended at the time of radical nephrectomy when RCC is suspected in children or young adults (Freedman et al, 1996; Asanuma et al, 1999; Geller et al, 2008; Selle et al, 2006; Bosquet et al, 2008). RCCs were traditionally thought to arise primarily from the proximal convoluted tubules, and this is probably true for the clear cell and papillary variants. However, it is now established that other histologic subtypes of RCC, such as chromophobe and collecting duct RCC, are derived from the more distal components of the nephron (Störkel, 1996; Oyasu, 1998; Pantuck et al, 2001a). The most generally accepted environmental risk factor for RCC is tobacco exposure, although the relative associated risks have been modest, ranging from 1.4 to 2.5 compared with controls (Table 49–5). All forms of tobacco use have been implicated, and risk increases with cumulative dose or pack-years (Kantor, 1977; La Vecchia et al, 1990; McLaughlin et al, 1995; McLaughlin and Lipworth, 2000; Moyad, 2001; Dhote et al, 2004; Lindblad, 2004; Lipworth et al, 2006; Carrizosa and Godley, 2009). Relative risk is directly related to duration of smoking and begins to fall after cessation, further supporting a cause-and-effect relationship (La Vecchia et al, 1990; McLaughlin et al, 1995; Parker et al, 2003b). Tobacco use accounts for 20% to 30% of cases of RCC in men and 10% to 20% in women (McLaughlin et al, 1995; McLaughlin and Lipworth, 2000). Table 49–5 Etiology and Environmental Factors of Malignant Renal Tumors Obesity is now accepted as another major risk factor for RCC with an increased relative risk of 1.07 for each unit of rising body mass index (Chow et al, 2000; Bergstrom et al, 2001; Bjorge et al, 2004; Calle and Kaaks, 2004; Reeves et al, 2007). The increased prevalence of obesity likely contributes to the increased incidence of RCC in Western countries, and it has been estimated that more than 40% of cases of RCC in the United States may be causally linked to obesity (Calle and Kaaks, 2004). Potential mechanisms linking obesity to RCC include lipid peroxidation leading to DNA adducts, increased insulin-like growth factor-1 expression, increased circulating estrogen levels, and increased arterionephrosclerosis and local inflammation (Kasiske et al, 1992; Huang et al, 1998; Gago-Dominguez et al, 2002; Calle and Kaaks, 2004). Hypertension appears to be the third major etiologic factor for RCC. Diuretics and other antihypertensive medications have also been implicated, but the weight of the epidemiologic evidence suggests that it is the underlying disorder, hypertension, rather than the treatment, that increases the risk of RCC (McLaughlin et al, 1995; Yuan et al, 1998; McLaughlin and Lipworth, 2000; Lipworth et al, 2006). The proposed mechanisms are hypertension-induced renal injury and inflammation or metabolic or functional changes in the renal tubules that may increase susceptibility to carcinogens (Lipworth et al, 2006). Although a number of other potential etiologic factors have been identified in animal models, including viruses, lead compounds, and more than 100 chemicals such as aromatic hydrocarbons, no specific agent has been definitively established as causative in human RCC (Bennington and Beckwith, 1947; Kantor, 1977). The potential role of trichloroethylene exposure has been actively investigated; some studies showed relative risks ranging from twofold to sixfold, but others have argued that inherent biases likely account for these results (Vamvakas et al, 2000; Mandel, 2001; Moyad, 2001; Bruning et al, 2003, Lipworth et al, 2006; Lock and Reed, 2006). However, Brauch and colleagues (1999) reported an increased incidence and unique pattern of von Hippel-Lindau gene (VHL) mutations in this population, which would argue in favor of a potential causative role for this compound. Slightly increased relative risks for RCC have been reported for workers in the metal, chemical, rubber, and printing industries and those exposed to asbestos or cadmium, but the data are not particularly convincing (Kolonel, 1976; Pesch et al, 2000; Moyad, 2001; Hu et al, 2002; Dhote et al, 2004; Lindblad, 2004; Lipworth et al, 2006; Carrizosa and Godley, 2009). Case-control studies have shown that RCC is more common among individuals with low socioeconomic status and urban background, although the causative factors have not been defined (Kantor, 1977; Goodman et al, 1986; Muscat et al, 1995; Yuan et al, 1998). The typical modern Western diet (high in fat and protein and low in fruits and vegetables), increased intake of dairy products, and increased consumption of coffee or tea have been associated with RCC, but the relative risks have been modest, and conflicting data are available in most instances (Yu et al, 1986; Lindblad et al, 1997; Moyad, 2001; Handa and Kreiger, 2002; Dhote et al, 2004; Lindblad, 2004; Murai and Oya, 2004, Lipworth et al, 2006; Wolk et al, 2006; Carrizosa and Godley, 2009). A family history of RCC may also be a factor; one study showed a relative risk of 2.9 for individuals with a first- or second-degree relative with RCC (Gago-Dominguez et al, 2001). Other potential iatrogenic causes include Thorotrast (which was used as a contrast agent in the past), and radiation therapy, but, again, the relative risks are low (Wenz, 1967; Romanenko et al, 2000). Vogelzang and colleagues (1998) reported four cases of RCC developing in a previously irradiated field, and a slightly increased incidence of RCC has been reported in men who received retroperitoneal irradiation for the treatment of testicular cancer. Survivors of childhood Wilms tumor also appear to be at increased risk for RCC, possibly related to prior radiation therapy or chemotherapy (Cherullo et al, 2001). An increased incidence of RCC is also observed in patients with end-stage renal failure and certain familial syndromes such as tuberous sclerosis, as discussed later (Ishikawa et al, 1990; Bjornsson et al, 1996; Neumann and Zbar, 1997). Since the early 1990s, significant advances have been made in our understanding of the molecular genetics of RCC. Novel familial syndromes of RCC have been identified, and the tumor suppressor genes and oncogenes contributing to the development of both sporadic and familial forms of this malignancy have been characterized (Table 49–6) (Linehan et al, 1995; Zbar et al, 1995; Schmidt et al, 1997; Weirich et al, 1998; Choyke et al, 2003; Linehan et al, 2003; Pavlovich et al, 2003; Zimmer and Iliopoulos, 2003; Pavlovich and Schmidt, 2004; Klatte and Pantuck, 2008; Nathanson and Stephenson, 2009). The impact of this new information should not be underestimated because it has fundamentally changed our perceptions about RCC. We now, more than ever, recognize the distinct nature of the various histologic subtypes of RCC, and advances in molecular genetics have contributed to a major revision in the histologic classification of this malignant neoplasm (Oyasu, 1998; Linehan et al, 2003; Young et al, 2008; Roma and Zhou, 2007; Pfaffenroth and Linehan, 2008; Zhou, 2009). A direct and beneficial impact on management of patients has also been achieved, with molecular targeted agents now extending survival for patients with advanced RCC (Linehan, 2002; Vira et al, 2007; Hudes et al, 2007, Motzer et al, 2007; Lane et al, 2007c; Kroog and Motzer, 2008; Clark and Cookson, 2008). Table 49–6 Familial Renal Cell Carcinoma (RCC) Syndromes * Also known as hybrid oncocytic tumors and containing features of both chromophobe RCC and oncocytoma. Data from Linehan, 2002; Choyke et al, 2003; Linehan et al, 2003; Maranchie and Linehan, 2003; Pavlovich et al, 2003; Pavlovich and Schmidt, 2004; Sudarshan and Linehan, 2006; Coleman, 2008; Pfaffenroth and Linehan, 2008; Hansel and Rini, 2008; and Nathanson and Stephenson, 2009. Knudson and Strong recognized that familial forms of cancer might hold the key to the identification of important regulatory elements known as tumor suppressor genes (Knudson, 1971; Knudson and Strong, 1972). Their observations about the childhood tumor retinoblastoma, in which familial cases tend to be multifocal and early onset, led them to propose a two-hit theory of carcinogenesis. They hypothesized that a gene product that could suppress tumor development must be involved and that both alleles of this “tumor suppressor gene” must be mutated or inactivated for tumorigenesis to occur. Furthermore, Knudson postulated that patients with the familial form of the cancer are born with one mutant allele and that all cells in that organ or tissue are at risk, accounting for the early onset and multifocal nature of the disease. In contrast, sporadic tumors develop only if a mutation occurs in both alleles within the same cell; and because each event occurs with low frequency, most tumors develop late in life and in a unifocal manner (Knudson, 1971; Knudson and Strong, 1972). Knudson’s hypothesis has proved true for retinoblastoma and a number of other tumor types, including RCC (Choyke et al, 2003; Linehan et al, 2003; Pavlovich et al, 2003; Zimmer and Iliopoulos, 2003; Pavlovich and Schmidt, 2004; Sudarshan and Linehan, 2006). Identification of familial cases of RCC was particularly important because it allowed linkage analysis between affected family members. The familial form of clear cell RCC is von Hippel-Lindau disease. This is a relatively rare autosomal dominant disorder that occurs with a frequency of 1 per 36,000 population. Major manifestations include the development of RCC, pheochromocytoma, retinal angiomas, and hemangioblastomas of the brainstem, cerebellum, or spinal cord (Table 49–7) (Horton et al, 1976; Go et al, 1984; Green, 1986; Jennings et al, 1988; Lamiell et al, 1989; Maher et al, 1990; Neumann and Zbar, 1997; Hansel and Rini, 2008; Nathanson and Stephenson, 2009). All of these tumor types are highly vascular and can lead to substantial morbidity, much of which can be avoided with prompt recognition and careful, skilled management. In particular, central nervous system lesions can lead to paralysis or death and the retinal lesions to blindness if they are not identified and managed in an expedient manner. Other common or important manifestations of von Hippel-Lindau disease include renal and pancreatic cysts, inner ear tumors, and papillary cystadenomas of the epididymis (Neumann and Zbar, 1997). An increased incidence of neuroendocrine tumors of the pancreas has also been reported in von Hippel-Lindau disease (Zbar et al, 1999; Coleman, 2008). Penetrance for all of these traits is far from complete, and some, such as pheochromocytomas, tend to be clustered only in certain families (Table 49–8) (Neumann and Zbar, 1997, Coleman, 2008; Nathanson and Stephenson, 2009). RCC develops in about 50% of patients with von Hippel-Lindau disease and is distinctive for its early age at onset (often in the third, fourth, or fifth decade of life) and for its bilateral and multifocal involvement (Horton et al, 1976; Go et al, 1984; Green, 1986; Jennings et al, 1988; Lamiell et al, 1989; Maher et al, 1990; Neumann and Zbar, 1997; Vira et al, 2007; Nathanson and Stephenson, 2009). With improved management of the central nervous system manifestations of the disease, RCC has now become the most common cause of mortality in patients with von Hippel-Lindau disease (Maher et al, 1990; Neumann and Zbar, 1997). Screening for von Hippel-Lindau disease and important considerations for the management of RCC in von Hippel-Lindau disease are reviewed later in this chapter. Table 49–7 Manifestations of the von Hippel-Lindau Syndrome Data from Horton et al, 1976; Green, 1986; Lamiell et al, 1989; Maher et al, 1990; Neumann and Zbar, 1997; Friedrich, 1999; Choyke et al, 2003; Linehan et al, 2003; Maranchie and Linehan, 2003; Pavlovich et al, 2003; Sudarashan and Linehan, 2006; Coleman, 2008; Hansel and Rini, 2008; and Nathanson and Stephenson, 2009. Early clues to the genetic elements involved in the development of RCC came from cytogenetics. These studies demonstrated a common loss of chromosome 3 in kidney cancer, particularly the clear cell variant, and led to intensive efforts to find a tumor suppressor gene in this region (Zbar et al, 1987; Seizinger et al, 1988; Hosoe et al, 1990; Lerman et al, 1991). Reports by Kovacs and colleagues (1989) and Cohen and associates (1979) of translocations involving chromosome 3 further implicated this chromosome as an important regulatory element. Southern blot testing and analysis for restriction fragment length polymorphisms with a wide variety of genetic markers subsequently demonstrated loss of heterozygosity in distinct regions on the short arm of chromosome 3 (reviewed by Jennings et al, 1995). Sophisticated molecular genetic linkage studies in patients with von Hippel-Lindau disease eventually led to the identification of the VHL tumor suppressor gene (Latif et al, 1993). This gene, which is located at chromosome 3p25-26, has now been completely sequenced, and its role as a tumor suppressor gene for both the sporadic and the familial forms of clear cell RCC has been confirmed (Gnarra et al, 1994; Linehan et al, 1995; Zbar, 1995; Furge and Teh, 2009; Nathanson and Stephenson, 2009). The VHL gene consists of three exons, and it encodes a protein of 213 amino acids. A large number of common mutations or “hot spots” in the gene have been identified, and a direct correlation between genotype and phenotype has been established in some cases (Gnarra et al, 1994; Linehan et al, 1995; Zbar, 1995; Brauch et al, 2000; Pavlovich and Schmidt, 2004; Kaelin, 2007). For instance, missense mutations (type 2 mutations) that result in a full-length but nonfunctional protein are commonly found in families with von Hippel-Lindau disease that develop pheochromocytomas, whereas deletions leading to a truncated protein (type 1 mutations) are typically found in families that do not develop pheochromocytomas (see Table 49–8) (Crossey et al, 1994; Linehan et al, 1995; Maher and Kaelin, 1997; Neumann and Zbar, 1997; Walther et al, 1999c; Hes et al, 2000; Friedrich, 2001; Pavlovich and Schmidt, 2004; Sudarshan and Linehan, 2006; Coleman, 2008). The identification of this tumor suppressor gene represented a major advance in the field and required close collaboration between clinical urologic-oncologists and molecular geneticists. The important historical steps in solving this challenging puzzle were reviewed by Linehan and colleagues (1995) and Zbar (1995), who spearheaded this important effort. As with most tumor suppressor genes, both alleles of the VHL gene must be mutated or inactivated for development of the disease; the observed inheritance patterns have conformed to Knudson’s hypotheses. As expected, almost all patients with von Hippel-Lindau disease were found to have germline mutations of one allele of the VHL tumor suppressor gene, and autosomal dominant inheritance from the affected parent was confirmed (Gnarra et al, 1994; Linehan et al, 1995, 2003). The second allele is commonly lost by gene or chromosome deletion (Zbar, 1995). Also, as predicted, most sporadic clear cell RCCs were found to harbor mutations or other genetic mechanisms, such as hypermethylation, that inactivated both alleles of the VHL gene (Zbar, 1995; Linehan et al, 2003; Nathanson and Stephenson, 2009). However, they differ in that both mutations must be acquired after birth, accounting for the late onset and the unifocal nature of the sporadic form of the disease. Subsequent work has focused on the function of the VHL protein and its potential mechanisms of action. The VHL protein is known to bind to elongins B and C, CUL-2, and RBX1 to form an E3 ubiquitin ligase complex and thereby modulates the degradation of important regulatory proteins (Gorospe et al, 1999; Lisztwan et al, 1999; Zbar et al, 1999; Wiesener et al, 2001; George and Kaelin, 2003; Linehan et al, 2003; Pavlovich and Schmidt, 2004; Sudarshan and Linehan, 2006; Vira et al, 2007). A critically important function of the VHL protein complex is to target the hypoxia-inducible factors 1 and 2 (HIF-1 and HIF-2) for ubiquitin-mediated degradation, keeping the levels of HIFs low under normal conditions. The HIFs are intracellular proteins that play an important role in regulating cellular responses to hypoxia, starvation, and other stresses. Inactivation or mutation of the VHL gene leads to dysregulated expression of the HIFs (Maxwell et al, 1999; Yu et al, 2001), and they begin to accumulate in the cell. This, in turn, leads to a severalfold upregulation of the expression of vascular endothelial growth factor (VEGF), the primary angiogenic growth factor in RCC, contributing to the pronounced neovascularity associated with clear cell RCC (Gnarra et al, 1996; Iliopoulos et al, 1996; Gunningham et al, 2001; Igarashi et al, 2002; Linehan et al, 2003; Sudarshan and Linehan, 2006; Vira et al, 2007). HIFs also upregulate the expression of tumor growth factor-α, platelet-derived growth factor (PDGF), glucose transporter (Glut 1), erythropoietin, and carbonic anhydrase IX (CA-IX), a tumor-associated antigen with specificity for clear cell RCC (Fig. 49–9) (Zbar et al, 1999; Wykoff et al, 2000; Turner et al, 2002; Wiesener et al, 2002; Linehan et al, 2003; Grabmaier et al, 2004; Sudarshan and Linehan, 2006; Vira et al, 2007). In addition, the VHL protein appears to influence the cell cycle, cellular differentiation, and intracellular processing of important matrix molecules such as fibronectin, and it may impact the metastatic process by upregulating the chemokine receptor CXCR4. All of these functions may contribute to the pathogenesis and distinctive character of this disease (Lieubeau-Teillet et al, 1998; Kamada et al, 2001; Bindra et al, 2002; Hergovich et al, 2003; Linehan et al, 2003; Na et al, 2003; Pavlovich and Schmidt, 2004; Klatte and Pantuck, 2008). (From Linehan WM, Walther MM, Zbar B. The genetic basis of cancer of the kidney. J Urol 2003;170:2163–72.) Other genetic elements potentially involved in the development of sporadic clear cell RCC include additional loci on the short arm of chromosome 3 and the TP53 and PTEN tumor suppressor genes. Sophisticated molecular analyses, including complete gene sequencing and assessment for inactivation of the promoter by hypermethylation, has failed to reveal VHL gene abnormalities in a small proportion of sporadic clear cell RCCs, and the search for additional regulatory elements has continued (Clifford et al, 1998b; Hamano et al, 2002; Banks et al, 2006). Loss of heterozygosity has also been observed at 3p12-p14 and 3p21.2-p21.3 and is particularly common in tumors with wild-type VHL status (Shridhar et al, 1997; van den Berg and Buys, 1997; Clifford et al, 1998a; Lott et al, 1998; Velickovic et al, 2001; Bodmer et al, 2002). The functional importance of these loci is suggested by experiments showing that the transfer of fragments of chromosome 3 containing only these genetic elements can suppress tumorigenesis in RCC cell lines (Van den Berg and Buys, 1997; Lovell et al, 1999; Bodmer et al, 2002). A candidate tumor suppressor gene at 3p12 has been described and may contribute to a VHL-independent pathway to RCC (Lovell et al, 1999). Increased immunostaining for TP53 has been reported in 6% to 40% of RCCs, with some studies suggesting a correlation with tumor grade and stage (Reiter et al, 1993; Uhlman et al, 1994; Haitel et al, 1999). However, the data regarding TP53 in RCC have been controversial, and no clear consensus is available at this time. Of more relevance is the PTEN tumor suppressor gene, which is downregulated in a subset of RCC tumors (Alimov et al, 1999; Kondo et al, 2001; Brenner et al, 2002; Velickovic et al, 2002; Horiguchi et al, 2003; Shin et al, 2003; Hara et al, 2004; Robb et al, 2007; Hager et al, 2007; Pantuck et al, 2007; Klatte and Pantuck, 2008). The PTEN protein inhibits phosphatidylinositol-3-kinase–dependent activation of protein kinase B (Akt), a key intermediary in the mammalian target of rapamycin (mTOR) pathway (Kim et al, 2009). Loss of PTEN leads to constitutive activation of mTOR, which promotes tumorigenesis, and this pathway has proven to be fertile ground for pharmacologic intervention (see Tumor Biology and Clinical Implications and Chapter 50) (Hudes et al, 2007; Klatte and Pantuck, 2008; Kim et al, 2009; Hudes, 2009b). Oncogenes, such as c-MYC, c-ERBB1, c-Ha-RAS, c-FOS, and RAF-1, have also been studied in clear cell RCC, but the available data suggest limited involvement (Slamon et al, 1984). Downregulation of DNA mismatch repair genes may contribute to genetic instability in RCC and allow accumulation of multiple genetic defects (Deguchi et al, 2003). Several studies have documented distinct cytogenetic findings in non–clear cell histiotypes of RCC; chromosome 3 and VHL gene abnormalities are uncommon in these variants (Störkel et al, 1997; Oyasu, 1998; Sudarshan and Linehan, 2006; Vira et al, 2007). These observations suggested a distinct genetic basis for non–clear cell RCC. Papillary RCC, the second most common histologic subtype of RCC, is characterized by trisomy for chromosomes 7 and 17 as well as abnormalities on chromosomes 1, 12, 16, 20, and Y (Störkel et al, 1997; Oyasu, 1998; Pavlovich et al, 2003). In 1995, Zbar and colleagues at the National Cancer Institute reported a second familial syndrome of RCC—hereditary papillary RCC (HPRCC). This followed a number of isolated case reports that suggested clustering of papillary RCCs within certain families (Zbar et al, 1994). In Zbar and colleagues’ series (1995) there were 10 families with 41 affected members (29 men and 12 women). Median age at diagnosis was 45 years, and most patients developed multifocal and bilateral papillary RCC. Type 1 papillary RCC is typically found in this syndrome rather than type 2, which is commonly seen in the hereditary leiomyomatosis and RCC syndrome. Unlike von Hippel-Lindau disease, most patients with HPRCC do not develop tumors in other organ systems (Czene and Hemminki, 2003; Pfaffenroth and Linehan, 2008; Nathanson and Stephenson, 2009). Mean survival in affected individuals was only 52 years in Zbar and colleagues’ series, although the number of patients dying of RCC was not defined. The development of CKD due to a combination of malignant replacement of the renal mass and loss of functioning nephrons secondary to various interventions is a potential contributor to morbidity and mortality in this syndrome (Ornstein et al, 2000). CT is the preferred imaging modality for patients with HPRCC because it has the greatest sensitivity for detecting the small, hypovascular lesions that are common in this syndrome (Sudarshan and Linehan, 2006; Pfaffenroth and Linehan, 2008; Nathanson and Stephenson, 2009). Studies of families with HPRCC demonstrate an autosomal dominant mode of transmission, similar to all of the familial RCC syndromes, and provide insight into the molecular genetics of HPRCC as well as a subset of patients with sporadic papillary RCC (Zbar et al, 1995; Schmidt et al, 1997; Linehan et al, 2003, Vira et al, 2007). Again, molecular linkage analysis in affected families played a key role in the discovery of this gene, which was localized to chromosome 7q31. However, in this case, the inciting event is activation of a proto-oncogene, rather than inactivation of a tumor suppressor gene. Missense mutations of the c-MET proto-oncogene at 7q31 were found to segregate with the disease, implicating it as the relevant genetic locus (Schmidt et al, 1997; Pavlovich et al, 2003). The protein product of this gene is the receptor tyrosine kinase for the hepatocyte growth factor (also known as scatter factor), and its activation leads to cellular proliferation and other potentially tumorigenic effects (Vira et al, 2007). Most of the mutations in HPRCC have been found in the tyrosine kinase domain of c-MET and apparently lead to constitutive activation (Schmidt et al, 1997; Pavlovich et al, 2003; Sudarshan and Linehan, 2006). The mutated MET protein can transform NIH 3T3 murine fibroblasts and is tumorigenic in immunodeficient murine models. Trisomy for chromosome 7, which is commonly found in HPRCC, develops primarily through duplication of the chromosome harboring the mutant allele of the c-MET proto-oncogene and effectively increases the dosage of the activated receptor (Zhuang et al, 1998; Sudarshan and Linehan, 2006; Hansel and Rini, 2008). Relatively early onset and multifocality in HPRCC are due to inheritance of the mutated c-MET gene, which places all the cells in the kidney at risk from birth, but the incomplete penetrance and variable clinical courses associated with this syndrome suggest that additional genetic loci or epigenetic phenomena may modulate the phenotype (Choyke et al, 2003; Linehan et al, 2003; Pavlovich et al, 2003; Pavlovich and Schmidt, 2004; Schmidt at al, 2004; Vira et al, 2007). Schmidt and colleagues (2004) have described three more families with HPRCC and have shown age-dependent penetrance and a variable clinical course according to the site of mutation and family involved. Whereas tumors in HPRCC tend to be less aggressive than their sporadic counterparts, it is clear that some can metastasize and become lethal (Schmidt et al, 2004; Vira et al, 2007). Schmidt and colleagues report c-MET mutations in 13% of patients with sporadic papillary RCC, suggesting that this molecular defect also contributes to a subset of this disease population (Schmidt et al, 1999; Sudarshan and Linehan, 2006; Vira et al, 2007). Small molecule inhibitors of the c-MET receptor are currently in development and may prove useful for the management of HPRCC and the subset of patients with sporadic RCC who harbor this mutation (Bellon et al, 2008; Hansel and Rini, 2008; Pfaffenroth et al, 2008). In 2001, Launonen and colleagues described a new familial renal cancer syndrome in which patients commonly develop cutaneous and uterine leiomyomas and type 2 papillary RCC (Choyke et al, 2003; Kiuru and Launonen, 2004; Pavlovich and Schmidt, 2004; Sudarshan and Linehan, 2006; Sudarshan et al, 2007). Mean age at diagnosis is in the early 40s (Hansel and Rini, 2008; Nathanson and Stephenson, 2009). Renal tumors in this syndrome are unusual for familial RCC in that they are often solitary and unilateral, and they are more likely to be aggressive than other forms of familial RCC (Linehan et al, 2003; Maranchie and Linehan, 2003; Pavlovich and Schmidt, 2004; Sudarshan and Linehan, 2006; Grubb et al, 2007; Hansel and Rini, 2008). Collecting duct RCC, another highly malignant variant of RCC, has also been observed in this syndrome, which was named hereditary leiomyomatosis and renal cell cancer (HLRCC) syndrome (Linehan et al, 2003; Grubb et al, 2007; Nathanson and Stephenson, 2009). The histologic hallmark of these tumors is large, prominent eosinophilic nuclei and nucleoli with perinucleolar clearing (Grubb et al, 2007; Merino et al, 2007). The HLRCC locus was mapped to a region on 1q42-44, and this was later shown to be the site of the fumarate hydratase gene (Tomlinson et al, 2002; Linehan et al, 2003; Toro et al, 2003; Pavlovich and Schmidt, 2004; Alam et al, 2005). Fumarate hydratase is an essential enzyme in the Krebs cycle of oxidative metabolism. Again, autosomal dominant inheritance was observed, and this appears to be a tumor suppressor gene rather than an oncogene (Linehan et al, 2003; Pavlovich and Schmidt, 2004). The mechanistic link between a metabolic enzyme located in the mitochondria and tumorigenesis is still an enigma and remains an active area of investigation (Pollard et al, 2003; Pavlovich and Schmidt, 2004). One hypothesis is that fumarate accumulation may stabilize HIF-1 by preventing its hydroxylation, which targets it for degradation (Isaacs et al, 2005; Sudarshan and Linehan, 2006; Ratcliffe, 2007; Coleman, 2008; Hansel and Rini, 2008; Pfaffenroth and Linehan, 2008). Penetrance for RCC in HLRCC is lower than for the cutaneous and uterine manifestations, with only a minority (20%) of patients developing RCC (Choyke et al, 2003; Sudarshan and Linehan, 2006; Pfaffenroth and Linehan, 2008; Nathanson and Stephenson, 2009). In contrast, almost all individuals with this syndrome will develop cutaneous leiomyomas and uterine fibroids (if female), usually manifesting at the age of 20 to 35 years (Sudarshan and Linehan, 2006
Historical Considerations
Classification
MALIGNANT
BENIGN
INFLAMMATORY
Radiographic Evaluation of Renal Masses
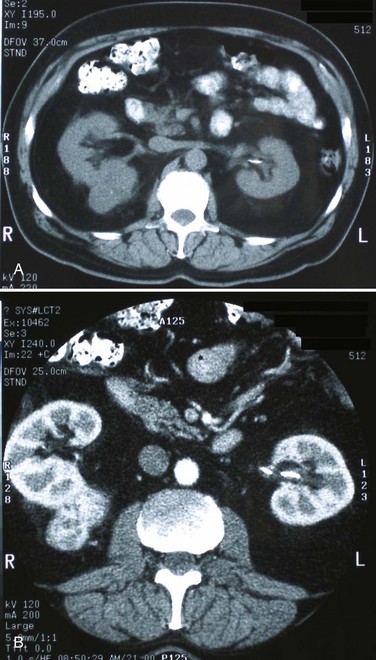
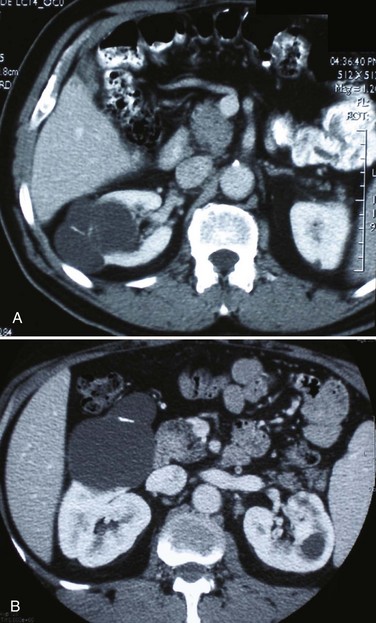
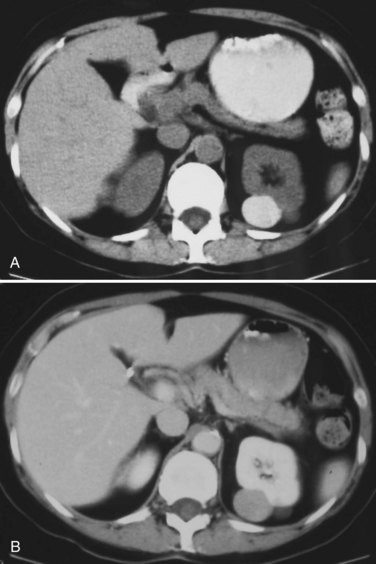
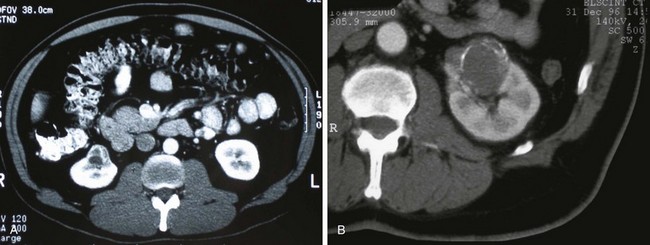
Renal Cell Carcinoma
Incidence
Etiology
Established
Putative
Familial Renal Cell Carcinoma and Molecular Genetics
SYNDROME
GENETIC ELEMENT
MAJOR CLINICAL MANIFESTATIONS
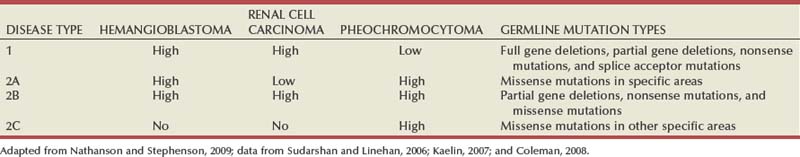
von Hippel-Lindau
VHL gene (chromosome 3p25-26)
Hereditary papillary RCC
c-MET proto-oncogene (chromosome 7q31)
Familial leiomyomatosis and RCC
Fumarate hydratase (chromosome 1q42)
Birt-Hogg-Dubé
BHD1 gene (chromosome 17p12q11)
von Hippel-Lindau Disease, VHL Gene, and Genetics of Clear Cell Renal Cell Carcinoma
ORGAN SYSTEM
LESION
INCIDENCE (%)
Eye
Benign retinal angiomas
49-59
Central nervous system
Benign hemangioblastomas
42-72
Kidney
Clear cell renal cell carcinoma
24-70
Renal cysts
22-59
Adrenal gland
Pheochromocytoma
18
Pancreas
Islet cell tumors
12
Malignant islet cell tumor
2
Pancreatic cysts
21-72
Epididymis
Cystadenoma
10-26
Ear
Endolymphatic sac tumor
10
Familial Papillary Renal Cell Carcinoma and Genetics of Papillary Renal Cell Carcinoma
Hereditary Leiomyomatosis and Renal Cell Carcinoma
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Prosthetic Surgery for Erectile Dysfunction
Prosthetic Surgery for Erectile Dysfunction
 Neuropathic Dysfunction of the Lower Urinary Tract
Neuropathic Dysfunction of the Lower Urinary Tract
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Malignant Renal Tumors