Chapter 96 Liver and pancreas transplantation immunobiology
Maintenance Minimization, Immunosuppressant Withdrawal, and Tolerance
Over the past 5 decades, the ability to manipulate the immune response has become increasingly selective and less morbid. Through a general understanding of alloimmunity and the unique properties of the liver and pancreas, transplantation results have steadily improved (U.S. Organ Procurement and Transplantation Network, 2009). This chapter provides an overview of the principles governing immune management of liver and pancreatic transplant recipients, highlighting agents and strategies generally available for clinical use.
Physiologic Immunity
The immune system is typically divided into two complementary arms, innate and acquired. The innate immune system recognizes general motifs that universally represent pathologic states, such as ischemia, necrosis, trauma, and nonhuman cell surfaces (Dempsey et al, 1996; Fearon & Locksley, 1996; Matzinger, 1994, 2001). The acquired immune system distinguishes specific pathogens through antigen presentation and recognition. Both systems interact to maintain overall homeostasis. Typically, innate responses localize acquired responses to sites of pathologic processes and are less overtly regulated. In contrast, acquired immune responses lead to carefully regulated destruction of antigen-expressing tissue. The regulatory checks on acquired immunity prevent autoimmunity and uncontrolled lymphocyte proliferation. It is important to recognize that the acquired immune system is tailored for the individual based on each person’s unique MHC makeup. Evolutionarily, this diversity reduces the chance that any single pathogen can evade all individuals within a population; however, this also means that one person’s acquired immune response may not respond appropriately when placed in the context of another person’s MHC.
Innate Immunity
Receptors of innate immunity are cell bound on macrophages, neutrophils, and natural killer cells, and they circulate freely in the form of complement (Dempsey et al, 1996; Fearon & Locksley, 1996; Wright et al, 1990). Innate immunity is limited in specificity, and it retains broad reactivity to lipopolysaccharide (LPS) and components of pathogenic organisms. Importantly, the receptors of innate immunity are conserved between individuals and, in general, function similarly in physiologic and transplant situations. Once activated, the innate immune system initiates cytolytic pathways and recruits the acquired immune response.
The complement cascade is the primary mediator of cytolysis, and the byproducts of complement, along with phagocytic cells, signal initiation of acquired immunity (Baldwin et al, 2010; Wasowska et al, 2007). Platelets also have been increasingly recognized as serving an innate immune role through the release of chemotactic proteins and other immunostimulatory molecules (Kirk et al, 2009). Professional APCs not only engulf cells coated with complement but also those bearing foreign carbohydrate moieties (Hart, 1997).
Toll-like receptors (TLRs) have also been recognized as important for innate APC activation. This highly conserved family of receptors binds to pathogen-associated molecular pattern motifs commonly expressed on invading pathogenic organisms (Akira & Takeda, 2004). Engulfed cells are processed by the APC into protein fragments and are expressed on the cell surface bound to MHC molecules. Subsequently, T cells that are specific for these peptides can recognize their cognate antigens and become activated. Interestingly, the TLRs expressed in the liver differ from those expressed in the periphery and tend to be less responsive to ambient LPS (Hart, 1997). This is likely an adaptation to portal bacteremia and is thought to make the liver more tolerant of minor perturbations that would evoke an innate response in other organs.
Acquired Immunity
Specific recognition is the hallmark of the acquired immune system. The lymphocyte receptors (T-cell receptor and antibody) have evolved to distinguish an extremely diverse group of antigens. Furthermore, antigen recognition induces physiologic changes in the recognizing cell that lowers its threshold for subsequent encounters and leads to the phenomenon of immunologic memory, the more rapid response to subsequent antigen encounters (Ahmed & Gray, 1996). T-cell receptors (TCRs) bind peptide antigens that have been processed and presented in combination with MHC, and B-cell immunoglobulins bind antigens in their native conformation at a site remote from the B cell.
Cellular Immunity
Formation of the TCR is fundamental to understanding its function (Cooper, 1987; Davis & Bjorkman, 1988). T cells are formed in the bone marrow and fetal liver and migrate to the thymus during development. After entering the thymus, T cells undergo rearrangement of the DNA that encodes the TCR (Gill & Gulley, 1994). Each gene rearrangement results in generation of a TCR with specificity restricted to one epitope or structurally similar epitopes. The sum of all random TCR gene rearrangements generates TCRs with approximately 109 specificities, essentially all possible combinations of MHC and peptide antigen; if these T cells were released into the periphery, they would mediate fatal autoimmunity. Accordingly, thymic selection eliminates those cells likely to evoke autoimmunity (Bevan, 1997; Kappler et al, 1987).
Thus all surviving T cells bind to self MHC. The developing T cells then move into the thymic medulla, where either CD4 or CD8 expression is lost. If binding to the self MHC in the medulla results in a high-affinity interaction, these T cells are also eliminated, a process known as negative selection. Therefore, the majority of cells released from the thymus bind to self MHC without becoming activated; however, autoreactive cells occasionally escape thymic selection and serve as the etiology of diseases such as sclerosing cholangitis (see Chapter 41), autoimmune hepatitis (see Chapter 97A), and type 1 diabetes (see Chapter 101); thus additional regulation is required to prevent autoimmunity. In fact, a single interaction of TCR and antigen-bearing MHC is inadequate to trigger T-cell activation; rather, approximately 8000 TCR-MHC interactions over a period of several hours are needed to initiate activation (Kumagai et al, 1987; Rothenberg, 1996; Viola & Lanzavecchia, 1996), which further limits the likelihood of autoimmunity. Costimulatory molecules markedly alter this need for redundancy, and we discuss this below.
Immune responses are also regulated by accessory cell surface molecules that limit the types of cells with which a T cell can interact (Leahy et al, 1992; Saizawa et al, 1987). Parenchymal cells of the body express class I MHC and display internal cellular peptides within the binding groove of this molecule. T cells charged with destruction of diseased or infected parenchymal cells express CD8 that stabilizes TCR ligation with class I MHC. These cells are termed cytotoxic T cells. CD8+ T-cell killing can occur through either Ca2+-dependent secretory mechanisms or Ca2+-independent direct cell-contact mechanisms (Berke, 1995).
Hematopoietic cells express class I and class II MHC. Class II MHC displays peptide fragments that have been phagocytized from the extracellular space (Germain, 1994; Monaco, 1993). CD4 stabilizes the TCR–class II MHC interaction. CD4+ cells interact with dendritic cells, macrophages, and in some cases activated endothelial cells that display antigen. In addition, the resting sinusoidal endothelial cells of the liver have the ability to present antigen to T cells, making the liver an organ with considerable ability to evoke or suppress an immune response (Knolle & Gerken, 2000). The interaction between CD4+ T cells and APCs produces APCs that have the ability to martial CD8+ T cells (Lanzavecchia, 1998; Ridge et al, 1998). This process is mediated through upregulation of APC cell surface molecules known as costimulation receptors, thus APCs initiate an immune response but require CD4+ T cells to activate the primary effector arm of the acquired immune system—the CD8+ T cells.
An additional subset of T cells, regulatory T cells (Treg), further control promiscuous immune responses. Regulatory T cells have the ability to suppress cytokine secretion, adhesion molecule expression, and costimulatory signaling. The most extensively studied population of Treg cells express CD4 and CD25, the high-affinity α-chain of the interleukin (IL)-2 receptor (Wood & Sakaguchi, 2003). Animal models have suggested that these cells play a critical role in controlling immune activation (Baecher-Allan et al, 2001; Wood & Sakaguchi, 2003). The prevailing evidence suggests that Treg is responsive to established inflammation, rather than serving a prophylactic role in preventing inflammation; however, harnessing the power of Treg to quell counteradaptive immune responses such as rejection is an ongoing area of research in autoimmunity and alloimmunity.
Humoral Immunity
B cells recognize antigen in its native, unprocessed form (Cambier et al, 1994). When antigen binds to two cell surface antibodies, the antibodies are brought together in a process known as cross-linking, which stimulates B-cell proliferation and differentiation into an antibody-secreting plasma cell. The activation threshold for a resting B cell is relatively high, as it is for T cells. As with TCR recognition, costimulation can lower this threshold substantially (Tedder et al, 1994). B cells also have the ability to internalize antigen bound to surface immunoglobulins and process them for presentation to T cells along with costimulation molecules (Lederman et al, 1992).
Antibody structure is determined in the bone marrow through mechanisms similar to those that govern the generation of TCR diversity in the thymus (Gill & Gulley, 1994; Hozumi & Tanegawa, 1976). Five different heavy-chain loci (µ, γ, α, ε, and δ) on chromosome 14 and two different light-chain loci (κ and λ) on chromosome 2—each with V, D and/or J, and C regions—are brought together randomly by the RAG-1 and RAG-2 apparatus to form a functional antigen receptor (Kim et al, 2000). The basic antibody structure consists of two identical heavy chains and two identical light chains. The type of heavy chain used dictates the immunoglobulin (Ig) type: IgM, IgG, IgA, IgE, or IgD. The overall structure of the antibody results in two identical antigen-binding sites and a common region, the Fc portion. Bound antibody triggers activation of the complement cascade (Baldwin et al, 1995). In addition, most phagocytic cells have receptors for the Fc portion of IgG, allowing them to actively engulf antibody-coated cells.
Unlike the TCR, B-cell immunoglobulin loci undergo alteration after B-cell stimulation to improve the functionality of the secreted antibody. Isotype switching is the process of shifting from the initial heavy-chain IgM to one of four types to improve function and specialization. IgG is the most significant soluble mediator of opsonization and is the dominant antibody produced in response to alloantigen. IgA is important in mucosal immunity, IgE is involved in mast cell–mediated immunity, and IgD is primarily cell bound. After a B cell is activated, the specific D and J regions of the used heavy- and light-chain genes undergo random alterations of the antigen binding site. The resultant B cell clones have altered antigen affinity, hence the term affinity maturation (Griffiths et al, 1984); those clones that have higher affinity for the target antigen have a selective survival advantage and form the basis for a more vigorous response on reexposure to the antigen.
Mediators of Context: Costimulation and Cytokines
Isolated TCR binding with an MHC-peptide complex or antibody ligation with an antigen is not usually sufficient for lymphocyte activation. Receptor-ligand pairs on T and B cells and APCs, known as costimulation receptors, determine the character of the T-cell response (Fig. 96.1; Allison & Krummel, 1995; Chambers & Allison, 1997; Crawford et al, 2006). The type of costimulatory signal received by the lymphocyte determines whether the cell will become activated, remain quiescent, die, or become resistant to subsequent immune stimulation.
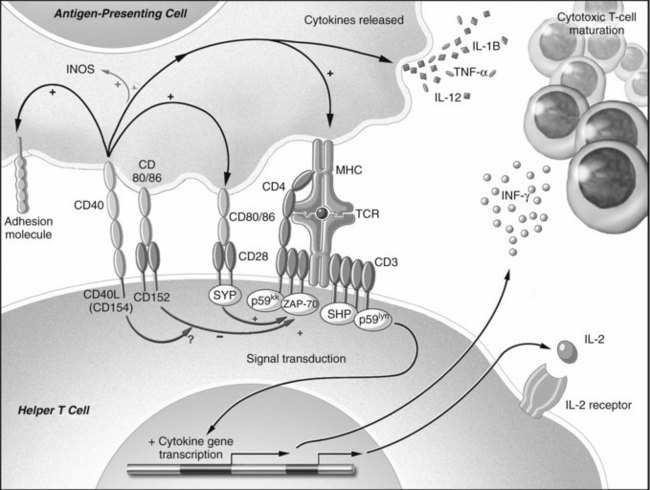
Although the mechanisms of costimulation have not been completely elucidated, it is known that binding of CD28 allows more efficient T-cell signal transduction. Through CD28-B7 interactions, the number of binding events required to trigger activation of a T-cell decrease from 8000 to 1500 (Rothenberg, 1996; Viola & Lanzavecchia, 1996). In contrast, when CTLA-4 binds B7, the T cell becomes incapable of producing IL-2 during the encounter and even in subsequent interactions (Blair et al, 1998). The CD19-CD21 complex provides comparable control of antigen receptor binding for B cells (Tedder et al, 1994).
Additionally, costimulation is mediated through another pair of receptors: CD40 found on dendritic cells, endothelium, B cells and other APCs, and CD154 on T cells and platelets (Armitage et al, 1992a, 1992b; Clark & Ledbetter, 1986; Noelle et al, 1992; Henn et al, 1998). The ability of APCs to stimulate a cytotoxic T-cell response is markedly augmented by the effects of CD40 binding. Following CD40 ligation, activating cytokines are released, and B7 molecules are upregulated (Bennett et al, 1998; Schoenberger et al, 1998). CD154 is upregulated after TCR ligation and provides positive feedback to the APC. In addition, CD154 is found in and released by activated platelets at sites of endothelial injury (Henn et al, 1998); therefore sites of trauma that recruit platelets create an environment of activating costimulatory molecules, thereby bridging the innate and acquired immune systems (Czapiga et al, 2004).
Direct cell-cell contact is not the only means by which immune cells communicate. Soluble mediators of communication known as cytokines, or interleukins, are polypeptides that are released from many cells; they can either activate or suppress adjacent cells (Arai et al, 1990), and the pattern of cytokine expression is thought to influence the resultant type of T-cell response (Mosmann et al, 1986; Mosmann, 1991). Once activated, T cells have been described by one of two cytokine-secretion phenotypes (see Chapter 10): cytotoxic T-cell responses are characterized by expression of interleukin (IL)-2, IL-12, IL-15, and interferon (IFN)-γ and are known as Th1 cells; T cells that promote humoral or eosinophilic responses are characterized by secretion of IL-4, IL-5, IL-10, and IL-13 and are known as Th2 cells.
Transplant Immunity
Most of the significant sequence polymorphism of MHC is located in the areas of the molecule that interact with the TCR, and individual variation in the sequence at the MHC-TCR interface defines alloreactivity. The lack of recipient T-cell thymic education with donor MHC leads to a nonphysiologically high frequency of alloreactive peripheral T cells. Many of these cells are crossreactive with antigen encountered during prior viral exposures, or even with autoantigens, as in the case of autoimmune disease. This is known as heterologous immunity, and it results in a situation whereby recipients have allospecific memory without having been exposed to the alloantigen (Adams et al, 2003). Thus a person’s immune response to a donor is the product of the individual’s MHC makeup and past immune exposures. This can lead to vigorous early rejection in apparently nonsensitized recipients.
T cells recognize alloantigen via their TCR in two distinct ways: either directly, by binding to donor MHC on transplanted tissues in the presence of donor costimulation, or indirectly, through self APC that has phagocytized and processed alloantigens to be presented, bound to self MHC and costimulation (Rogers & Lechler, 2001). In the case of transplanted organs, surgical trauma and ischemia exacerbate the potential for T-cell activation by causing class I and class II MHC upregulation (Gerritsen & Bloor, 1993). In addition, adhesion molecules and costimulation molecules are upregulated perioperatively (Takada et al, 1997; Hoffmann et al, 2002).
Initial T-cell binding to donor cells is nonspecific, mediated by adhesion molecules upregulated during donor cell activation (Fuggle & Koo, 1998). CD40 on donor APCs and endothelial cells is important in mediating cell activation in this setting via CD154 on T cells and activated platelets (Henn et al, 1998). Following nonspecific adhesion, MHC recognition occurs in the relatively high costimulation environment induced by surgical trauma and ischemia (Takada et al, 1997). Once alloreactive T cells are activated, they secrete cytokines, including IL-2 and IFN-γ, and they stimulate APCs to secrete IL-12 (Arai et al, 1990; Kirk et al, 1995; Krams et al, 1992). The resultant cytokine milieu recruits more T cells to the site of injury and potentiates clonal expansion. Secretory and cell contact–dependent mechanisms, perforin/granzyme and Fas mechanisms, respectively, are involved in T-cell cytotoxicity within the graft, resulting in graft destruction (Strehlau et al, 1997). Although acute rejection is the result of T-cell activation, antibody responses accompany many episodes. Cellular and soluble components of immunity mediate multiple distinct clinical rejection syndromes through cytokine mediated toxicity, cellular cytotoxicity, and direct effects of antibody and complement.
Clinical Rejection Syndromes
Hyperacute Rejection
Hyperacute rejection (HAR) is caused by donor-specific antibody that exists at the time of transplantation as a result of prior exposure to donor antigens or to antigens with crossreactivity. It develops precipitously within minutes to hours after graft reperfusion. Typically, HAR is avoided by confirming ABO compatibility and performing a crossmatch using techniques that detect donor-specific antibodies. When clinically relevant donor-specific antibodies are detected, graft survival is significantly decreased for most organ types (Noreen et al, 2003); however, the liver has long been regarded as relatively resistant to HAR, and crossmatching is often done only retrospectively (Neumann et al, 2001). Actually, the rates of acute rejection and long-term liver survival are similar among groups with a positive or negative crossmatch; this does not hold true for ABO-incompatible liver allografts (Egawa et al, 2004).
The role of antibody in liver transplantation remains an area of investigation at present. Unfortunately, the pancreas does not have similar resistance to HAR, and a positive crossmatch represents an absolute contraindication to pancreas transplantation. Although high titer antibodies mediate rapid graft injury, chronic exposure to lower titer antibody results in indolent graft damage, particularly in the case of the pancreas. The role of chronic alloantibody in liver transplantation remains controversial but likely mediates some degree of graft injury over time. Autoantibody directed against nonpolymorphic determinants of a graft is increasingly being recognized as detrimental in pancreas transplantation with recurrent β cell–specific autoimmunity responsible for at least some late graft loss after pancreas transplantation (Vendrame et al, 2010). Recurrent autoimmunity is also relevant in autoimmune hepatitis (Hytiroglou et al, 2009).
Acute Rejection
Acute rejection is most common between 4 days and 6 months after transplantation, and rejection that occurs during this time is considered early acute rejection; acute rejection after 6 months is considered late acute rejection. Liver allografts undergo acute rejection at a rate of approximately 24% to 47% (Fisher et al, 2004; Neuhaus et al, 2002; Wiesner et al, 2001
Related posts:
 Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
 Portal hypertension in children
Portal hypertension in children
 Liver transplantation for cholangiocarcinoma and other neoplastic diseases
Liver transplantation for cholangiocarcinoma and other neoplastic diseases
 Distal splenorenal shunt
Distal splenorenal shunt
 Interventional endoscopy: Technical aspects
Interventional endoscopy: Technical aspects
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


