in elaboration of renin and aldosterone in response to volume depletion. Both hypokalemia and nonrenal metabolic acidosis increase renal NH4+ synthesis, which increases urinary buffering capacity and urinary pH. The presence of NH4+ in the urine can be used clinically to distinguish hyperchloremic metabolic acidosis due to diarrhea from renal tubular acidosis (Table 18.2). In the latter, NH4+ excretion is invariably low. Urine pH, although a time-honored method to distinguish these disorders, is less reliable because urinary ammonium excretion is augmented in chronic metabolic acidosis due to diarrhea, causing urine pH to increase over time.
TABLE 18.1 Differential Diagnosis of Nongap Acidosis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 18.2 Diagnostic Criteria for Causes of Nongap Acidosis | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
principle defect in advanced renal failure is impaired ammoniagenesis and ammonium excretion.5 The latter is a result of impaired medullary ammonium transport and trapping of NH3/NH4+ in the outer and inner medulla.
TABLE 18.3 Clinical Application of Urine Anion Gap to Approximate Urine Ammonium Excretion | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
TABLE 18.4 Urine Osmolar Gap to Approximate Urine Ammonium Concentration | |||
|---|---|---|---|
|
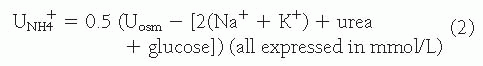
each day, 80% to 90% is reabsorbed in the proximal tubule. Effective HCO3– absorption in the proximal tubule is mediated by H+ secretion. Even though the distal tubule is responsible for the secretion of 50 to 80 mEq of H+ and final acidification of the urine, the vast majority of H+ secretion obviously occurs in conjunction with HCO3+ reclamation in the proximal tubule.
symporter or NBCe1, the enzymes carbonic anhydrase type II or IV, and the Na+/H+ exchanger (NHE-3) (Fig. 18.1).
TABLE 18.5 Etiology of Proximal Renal Tubular Acidosis (Type 2) with or without Fanconi Syndrome | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
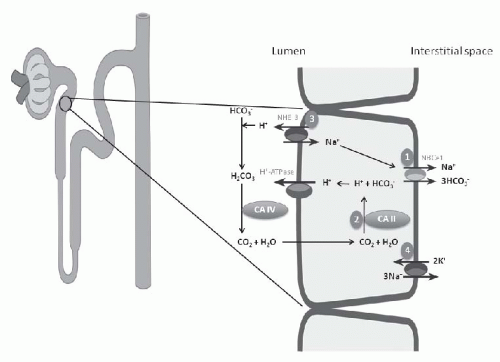 FIGURE 18.1 Pathogenesis of proximal renal tubular acidosis. Model of bicarbonate reabsorption in the proximal tubule showing described inherited defects of genes encoding proximal transport proteins that cause proximal renal tubular acidosis. 1, defect of basolateral Na+-HCO3– cotransporter; 2, defect of carbonic anhydrase type 2; 3, defect of Na+/H+ exchanger; 4, defect of Na+,K+ ATPase. See text for detail. |
coupled to apical sodium reabsorption. Fanconi syndrome is also associated with distal nephron dysfunction in some patients. The mechanisms involved are not known, but the evidence suggests that there may be a defect in Na+,K+-ATPase throughout the nephron.
proximal RTA, presumably as a result of enhanced calcium absorption in the distal nephron in response to increased bicarbonate delivery. Rickets, a frequent manifestation of the Fanconi syndrome, is a result of phosphate wasting, not proximal RTA or acidosis. Of particular concern in children with proximal RTA is growth retardation, a direct consequence of acidosis. Because growth retardation will correct with alkali therapy, this complication becomes one of the major indications for correction of the serum bicarbonate concentration.
TABLE 18.6 Treatment of Proximal Renal Tubular Acidosis | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
in the distal nephron and together are responsible for H+ secretion42 (Fig. 18.2). Immunohistochemical studies have localized the H+-ATPase to the apical membrane of acid secreting cells (type A intercalated cells) in the CCT and OMCT. Both the gastric and colonic H+,K+-ATPase subunits are expressed in intercalated cells of the cortical collecting duct and outer medullary collecting duct.42 These two isoforms of H+,K+-ATPase have been designated as the HK1 (“gastric”) and HKα2 (“colonic”) subunits. HKα1 is identical to the H+,K+-ATPase in gastric parietal cells, whereas HKα2 is homologous to the H+,K+-ATPase in distal colon. Several studies have demonstrated that HKα2 mRNA and protein (but not HKα1) are dramatically upregulated by chronic hypokalemia and chronic acidosis.43 Furthermore, increased H+,K+-ATPase activity in the outer and inner medullary collecting duct results in enhanced HCO3– absorption.44
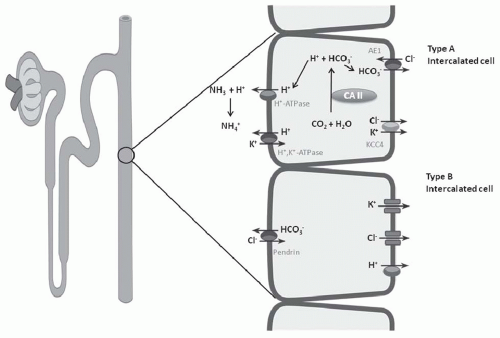 FIGURE 18.2 Type A and B intercalated cells of collecting duct. See text for detail. |
discussed in detail in Chapter 6, “new bicarbonate” restores the HCO3– lost in the ECF from buffering the acid products of metabolism.
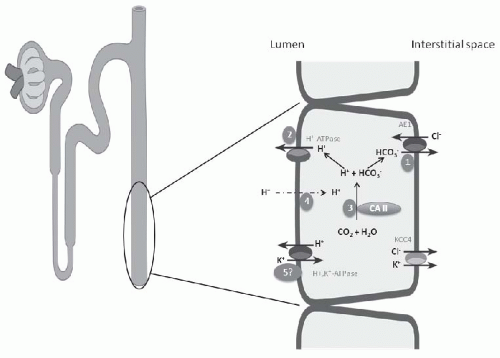 FIGURE 18.3 Pathogenesis of distal renal tubular acidosis (RTA). Model of type A intercalated cell in medullary collecting duct showing described inherited defects encoding distal transport proteins that cause classical distal RTA. 1, defect of basolateral HCO3–/Cl– exchanger; 2, defect of specific subunits of H+-ATPase; 3, carbonic anhydrase II deficiency; 4, backleak of H+ or gradient lesion (amphotericin B and presumed rare inherited abnormalities); 5, abnormality of H+,K+-ATPase (not verified, but may explain endemic [Northeastern Thailand] distal RTA with severe hypokalemia). |
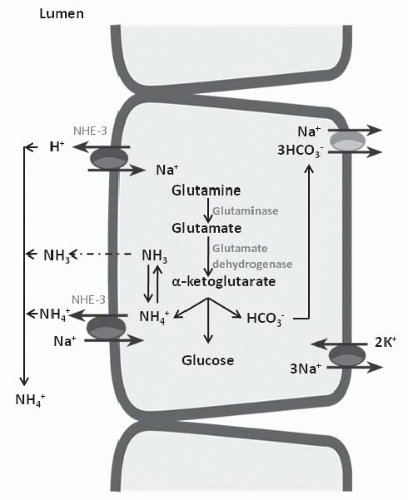 FIGURE 18.4 Ammoniagenesis and transport of ammonia/ ammonium in the proximal tubule. |
electrogenic H+-ATPase, the electroneutral H+,K+-ATPase on the apical membrane, and the HCO3–/Cl– exchanger on the basolateral membrane (Fig. 18.6). Alteration in the negative transepithelial potential difference, which is dependent on the rate of Na+ absorption, has a significant secondary impact on proton secretion by the electrogenic H+-ATPase. Thus, a decline in Na+ delivery or Na+ acidity through either impairment of epithelial Na+ channel (ENaC) function or through absence of mineralocorticoid will secondarily impair H+ secretion. A defect in H+ secretion in the CCT in response to a decline in Na+-transport dependent transepithelial voltage has been termed a “voltage defect.” Mineralocorticoids have been demonstrated to be a potent determinant of proton secretion. In the CCT, mineralocorticoids stimulate Na+ absorption (ENaC) increasing the lumen negative transepithelial potential, which stimulates electrogenic proton secretion secondarily.56 This early effect of aldosterone on ENaC is reinforced after several hours to upregulate the basolateral Na+,K+-ATPase as well. Taken together, mineralocorticoid increases the negative transepithelial potential, thus enhancing Na+ absorption. Mineralocorticoids have also been shown to stimulate the H+-ATPase in the cortical, outer, and inner medullary collecting tubules in the absence of Na+.40,57 Thus, in summary, both mineralocorticoids and Na+ absorption in the CCT have important regulatory effects on net H+ secretion in the collecting duct.
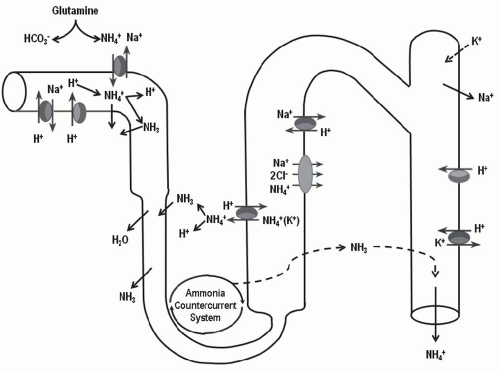 FIGURE 18.5 Summary of ammonia/ammonium transport pathways in the nephron. Possible defective ammoniagenesis and/or ammonium transport associated with distal renal tubular acidosis, discussed in text in detail. |
The occurrence of hypokalemia demonstrates that generalized CCT dysfunction or aldosterone deficiency is not causative. Initially the cause of classical hypokalemic DRTA was considered to be a “gradient lesion.” Alternatively, a defect in proton secretion is the most widely accepted explanation for the inability to maximally acidify the urine,59 in the majority of forms of classical DRTA (Fig. 18.3). The most notable exception of this mechanism is the defect induced by amphotericin B nephrotoxicity, for which insertion of a leak pathway in the apical membrane of the distal tubule by the antibiotic is causative (Fig. 18.3, number 4). An important feature in the determination of the pathogenesis of the acidification defect in these patients is the response of the urine PCO2 to NaHCO3 infusion. Infusion of NaHCO3 to produce a high HCO3– excretion rate results normally in distal nephron hydrogen secretion and the generation of a high CO2 tension in the urine. The magnitude of the urinary PCO2 (referred to as the urine-minus-blood PCO2, or U-B PCO2) is quantitatively related to distal nephron hydrogen ion secretion.38,60 A decrease in the rate of hydrogen ion secretion as a result of a defect of one of the H+ transporters on the apical membrane (H+– or H+,K+-ATPase) or the basolateral HCO3–/Cl– exchanger will lead to a low U-B PCO2. In contrast, a backleak of H+, as occurs with a “gradient” defect, has been shown in experimental models to be associated with a normal U-B PCO2. In patients with classical hypokalemic DRTA, the U-B PCO2 is usually subnormal, except in amphotericin B-induced DRTA.60,61,62 This finding supports the view that most patients with DRTA have a “rate” or “pump” defect (Fig. 18.3, number 1).
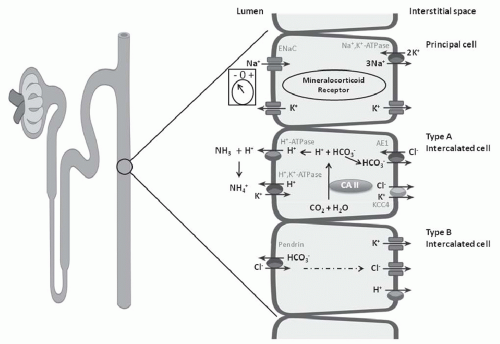 FIGURE 18.6 Three cell types in the collecting tubule: principal cell, and type A and type B intercalated cell. Mechanisms of transport discussed in text. |
investigators have described autosomal dominant mutations in the ATP6V1B1 gene encoding the B-subunit of the H+-ATPase in the kidney and cochlea, and these mutations are associated with sensorineural deafness. Another form of classical DRTA, inherited as an autosomal recessive defect, is associated with normal hearing and has been shown recently to be a mutation in the ATP6V0A4 gene that encodes for the A-subunit of this transporter.63,64,65
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


