GENE IDENTIFICATION
The basis of any inherited disease is an underlying alteration in genomic DNA that is transmitted from parent to offspring. Theoretically, one could compare the entire genomic sequence of an individual affected with a particular disease to that of unaffected individuals in order to identify the pathogenic difference. As simple as this approach sounds, it has, until recently, been a daunting challenge due to the size of the diploid human genome (6 × 109 base pairs [bp]), the limited throughput of traditional DNA sequencing methods (106 to 107 bp per day), and the high degree of normal variation in human populations. Important technical and bioinformatic developments have greatly changed the genetic disease discovery landscape, however. Whole genome sequencing is now a reality, and the <$ 1,000 genome will soon be here. Enigmatic clinical disorders that were once deemed too difficult to study using conventional positional cloning approaches because of their rarity are now giving up their secrets. In this section, we will briefly review the history of renal disease gene discovery, highlighting some of the most illustrative examples, and then discuss the impact of next generation sequencing on this field.
Prior to the easy availability of inexpensive DNA sequencing, multiple approaches had been developed to facilitate disease gene discovery. With some disease entities, a broader understanding of underlying pathogenic mechanisms had enabled the identification of potential “candidate genes.” Alport syndrome is an example of the successful application of this approach.
1 Biochemical analysis of the Alport glomerular basement membrane (GBM) identified a set of α chains of type IV collagen that were missing.
2,
3,
4,
5 Molecular techniques were then used to clone the type IV collagen genes, and mutation analyses revealed that sequence variants in a subset of these genes segregated with the disease. In a similar manner, the recognition that individuals suffering from the infantile form of Bartter syndrome have a clinical presentation similar to that of patients on loop diuretics prompted investigators to evaluate the drug’s target, the Na-K-2Cl cotransporter (
SLC12A1), as a probable candidate gene. As predicted for this recessive disease, inactivating mutations were found in both alleles in a subset of families.
6For other disorders, the underlying gene defect was identified by
expression cloning of candidate genes. In this approach, one identifies genes responsible for a particular function by the transfer of genetic material into cells that lack that function and then screening for activity. Typically, multiple pools of genes are used for the initial screening and then the search is focused on only those pools that demonstrate the desired activity. By using a reiterative process of serial dilutions and functional testing, one can ultimately identify the gene or genes responsible for the observed activity. Finally, the cloned candidate genes are scanned for sequence differences (mutations) that segregate with disease. This approach has been termed expression cloning and has been used most successfully to identify various transporters. The genes implicated in cystinuria,
SLC3A1 and
SLC7A9, were identified by this approach.
7,
8,
9,
10,
11 Likewise, the three subunits that comprise the epithelial sodium channel, ENaC, were isolated in this manner.
12 Inactivating mutations of each of the subunits have been associated with recessive forms of pseudohypoaldosteronism type I,
13,
14 whereas activating mutations of either the
β or γ subunit have been found in Liddle syndrome, an autosomal dominant form of hypertension.
15,
16Each of the prior approaches requires an in-depth understanding of the underlying pathobiology of the disease for its success. Unfortunately, we lack this information for most diseases. This necessitated the use of a strictly molecular genetic approach, termed
positional cloning, which seeks to identify a disease gene solely on the basis of its chromosomal location.
17In some cases, important positional clues were provided by a cytogenetic analysis of affected individuals. Several of the loci involved in the origin of renal tumors were identified by this approach (
Table 14.4). The gene responsible for the major form of Wilms tumor,
WT1, was initially discovered because of its involvement in WAGR syndrome (
Wilms tumor,
aniridia,
genitourinary anomalies, and mental
retardation).
18,
19,
20 Individuals affected by this disorder were found to have constitutional deletions of 11p13 involving a zinc finger transcription factor,
WT1, a paired box transcription factor (
Pax6), and adjacent DNA sequences. Fine mapping proved that
WT1 was responsible for the genetic susceptibility to Wilms tumor, whereas
Pax6 was responsible for the aniridia phenotype.
21 One of the familial forms of nonpapillary renal cell carcinoma (RCC) also was identified on the basis of its underlying chromosomal rearrangement. A translocation between chromosomes 3p and 8q (t[3;8][p14.2;q24.1]) was found to segregate with renal cell carcinoma.
22 Nearly 2 decades later, investigators identified the genes disrupted by the translocation. They determined that the chromosomal rearrangement resulted in a novel gene that consisted of 5′ elements of a gene called
FHIT (fragile histidine triad gene) fused to the coding sequence of
TRC8.23 The protein product of
TRC8 has high homology to the basal cell carcinoma/ segment polarity gene product, Patched, a signaling receptor, suggesting that it may have a similar function. Since that time, 11 further chromosome 3 translocations have been associated with a susceptibility to RCC, including several that disrupt candidate tumor suppressor genes such as
LSAMP and
NORE1. 24Perhaps one of the most striking examples of the power of cytogenetic abnormalities to expedite gene discovery is that provided by the search for
PKD1, the gene responsible for the most common form of ADPKD. A combination of molecular and genetic techniques had rapidly localized the gene to a 500 kilobase (kb) gene-rich segment, but the lack of known chromosome rearrangements or deletions coupled with the large number of potential candidate genes greatly complicated the search.
25,
26 Several years of mutation screening had failed to determine which one of the many candidates was in fact
PKD1 when an astute clinician identified an unusual family that had individuals with classic ADPKD as well as a child with both tuberous sclerosis and renal cysts. Because it was known that a major form of tuberous sclerosis (TSC2) was located near the
PKD1 gene,
27 cytogenetic studies of the family were undertaken. This revealed two individuals in the family with balanced translocations between chromosomes 16 and 22 (t[16;22] [p13.3;q11.21]).
28 The child with TSC2 had an unbalanced karyotype and was missing a portion of chromosome 22 as well as the telomeric portion of chromosome 16 (45XY/-16-22 + der[16][16qter-16p13.3::22q11.21-22qter]). It was correctly speculated that the
TSC2 gene was located in the portion of chromosome 16 that was lost while
PKD1 was likely to be the gene bisected by the translocation breakpoint. This was confirmed by additional studies and resulted in the identification of both
TSC2 and
PKD1.
28,
29Although chromosomal rearrangements are incredibly helpful when associated with disease, they are uncommon. Therefore, most gene searches began using linkage-based methods. Linkage analysis requires both well-characterized pedigrees and an array of genetic markers. Genetic markers are DNA variants that differ within the normal population in their length or sequence and can be used to trace inheritance of parental chromosomes within families. The principle underlying this approach is as follows: a genetic disease is assumed to be the clinical manifestation of a DNA mutation. Therefore, one can identify the location of the mutant gene by comparing the segregation of the disease phenotype with a battery of genetic markers. Alleles of loci (a specific chromosomal address of a DNA segment) on different chromosomes will appear to segregate randomly, whereas alleles of loci physically close on the same chromosome are inherited together and are linked. Meiotic recombination produces novel haplotypes by exchanging alleles between homologous chromosomes and the frequency with which this occurs depends in part on the distance separating them. In other words, if the alleles of two genes are adjacent to each other on the same chromosome segment, there will be no recombination between them. Statistical programs are used to score the probability that an observed association has not happened by chance. The most common approach determines the ratio of the probability of the observed associations assuming linkage to that of no linkage. The LOD score is the decimal logarithm of this ratio and is considered significant when greater than 3.
Until recently, it was necessary to clone the chromosomal interval in question, identify its genes, and then screen them for mutations. These steps often took many years to complete. The human genome project revolutionized this process by virtually providing the complete, annotated sequence of the human genome in public databases and producing dense genetic maps that included over a million single nucleotide polymorphisms (SNPs). SNP-chips (which query hundreds of thousands of SNPs in a single hybridization) have facilitated rapid genetic localization of disease loci, and the genomic maps have provided a comprehensive set of all potential candidate genes. Despite these advances, the task of identifying a disease-associated gene still remained a time-consuming endeavor prior to the widespread availability of high-throughput sequencing. The reason for this is that the resolution of genetic mapping is typically on the order of 500,000 to 1,000,000 bp. With an average gene density of one gene per 30,000 bp in gene-dense regions, a segment of this length could harbor between 30 to 40 genes! For diseases that are uncommon, the small number of family members available for testing often limited the resolution of genetic mapping to an area millions of base pairs in length.
Several shortcuts were used to minimize the level of effort. The first strategy was to use publicly available databases to determine the identity and likely function of genes within one’s target region. One could use a modest understanding of the pathobiology of a disease to narrow the field of candidates to a set whose functions were consistent with the underlying defect. For example, linkage studies revealed that one of the loci responsible for hereditary papillary renal carcinoma (HPRC) mapped to an interval on 7q31 that included the
MET proto-oncogene. The well-established relationship between
MET and other carcinomas prompted the investigators to focus their search on this gene, leading to the rapid discovery of pathogenic mutations.
30 In another example, Dent disease was known to be an X-linked disorder often associated with microdeletions of Xp11.
22,
31 Fisher et al.
32 initiated their search for the gene responsible for Dent disease by screening for expressed sequences that were encoded by the deleted segment. They identified a novel chloride channel family member that was deleted in many patients with Dent disease, that had a restricted pattern of expression, and whose function was consistent with the pathophysiology of the disease. They subsequently showed that mutations of
CLCN5 were responsible for this disease.
33 In the case of ADPKD, investigators seeking the identity of
PKD2 determined that one of the candidate genes in their genetic interval had homology to the gene product of
PKD1, polycystin-1. This gene was an obvious candidate for
PKD2, and mutation analysis quickly confirmed this suspicion.
34One of the most interesting examples of this approach was its use to identify a novel locus for Bardet-Biedl syndrome (BBS). Investigators had determined that this rare, autosomal recessive disorder, which is characterized by obesity, mental retardation, anosmia, fibrocystic renal disease, congenital hepatic fibrosis, and left/right axis defects, was genetically heterogeneous. A number of loci were known, and investigators had found that their respective gene products were localized to either the basal body and/or the primary cilium. The overlap in clinical features between BBS and other diseases that result from dysfunction of ciliary proteins gave rise to the idea that candidate genes for BBS and other similar diseases could be identified based on this property. Scientists compared the complete genomic sequence of multiple species that have cilia to those that do not, and identified a set of genes exclusively present in organisms with cilia and basal bodies. Two of the genes mapped to a previously defined interval for
BBS5 that contained 230 predicted genes. Molecular testing identified mutations in one of the two genes in BBS5 families.
35 The locus encodes a novel protein of unknown function that would have otherwise been a low priority candidate for further study.
A second approach that had been used to speed the pace of gene discovery was to search for disease-associated microscopic chromosomal abnormalities that were below the level of resolution of standard cytogenetic analyses. The genes responsible for the most common form of nephronophthisis (
NPHP1) and for cystinosis (
CTNS) were identified in this manner. In the case of
NPHP1, large-scale rearrangements were detected in 80% of the patients belonging to inbred or multiplex
NPHP1 families and in 65% of the sporadic cases.
36 Most of the time, large homozygous deletions of approximately 250 kb involving a 100-kb inverted duplication were discovered to disrupt the gene. In a small number of individuals, oligo-base pair mutations were identified, proving that
NPHP1 was the specific gene responsible for the disorder.
37,
38 CTNS was identified in a similar manner. Investigators found that one of the genetic markers used in their study was homozygously deleted in 23 out of 70 patients.
39 They quickly focused their search on the minimal deleted region and identified a ubiquitously expressed transcript that was disrupted in all patients with deletions involving this segment. They subsequently found single or oligo-base pair mutations in many of the remaining patients, thus proving that this gene and not one of its neighbors was in fact responsible for the disease.
A third strategy had been to determine the expression pattern of the various candidates and see if any were consistent with the clinical features of the disorder. Fuchshuber and colleagues
40 had localized a form of steroid-resistant idiopathic nephrotic syndrome (NPHS2) to a 2.5 million-base pair interval on chromosome 1. They had identified multiple putative candidates but focused their search on one whose expression by Northern blot was detectable only in fetal and adult renal tissues. They subsequently discovered recessive, inactivating mutations in
NPHS2, and further showed that its expression was restricted to glomerular podocytes.
41 In a similar manner, the kidney-restricted pattern of expression of
PCLN1 helped to identify it as a probable candidate gene for primary hypomagnesemia.
42In a number of diseases, gene discovery resulted more from good luck than from the pursuit of a particular strategy. The ability to manipulate the murine genome through gene targeting (described in more detail later) has allowed investigators to generate a lengthy list of murine models of human diseases. In most cases, scientists first identified the
disease gene and then created a mutant phenotype in the mouse with the intention of modeling the human disease state (see the subsequent text). In some cases, however, the genes targeted for study had not been previously implicated in a genetic disorder; rather, they had been selected for study because the investigators had a fundamental interest in their biologic properties. Careful analysis of the murine phenotypes revealed surprising similarity to human diseases, leading investigators to test for mutations in their human homologues.
It was in this way that
LMX1B, which encodes LIM homeobox transcription factor 1β, was found mutated in Nail-Patella syndrome (NPS). Investigators with an interest in basic developmental processes had targeted this gene for inactivation and discovered limb and kidney defects in
Lmx1b mutant mice that were remarkably similar to those observed in human NPS.
43 They quickly identified three independent NPS patients with de novo heterozygous mutations of
LMX1B.44 The identification of
CD2AP (CD2-associated protein) as a cause of steroid-resistant nephrotic syndrome is another example.
45,
46 CD2AP was thought to be an adapter protein critical for stabilizing contacts between T cells and antigen presenting cells. Mice that were null for
Cd2ap had compromised immune function but died unexpectedly at 6 to 7 weeks from renal failure. The investigators showed that homozygotes developed proteinuria associated with defects in epithelial cell foot processes and eventual glomerulosclerosis. CD2AP was found expressed in podocytes where it associates with nephrin, the primary component of the slit diaphragm. Subsequent studies in humans discovered
CD2AP mutations in patients with focal segmental glomerulosclerosis.
47,
48,
49It is likely that additional fortuitous relationships will be established, as the list of murine genes that are inactivated by gene targeting becomes more complete. The National Institutes of Health (NIH)-sponsored Knockout Mouse Project (http://www.komp.org/), in collaboration with the International Knockout Mouse Consortium (http://www .knockoutmouse.org/), is aiming to mutate all protein-coding genes in the mouse and then perform broad, standardized phenotyping on a large subset (https://commonfund.nih. gov/KOMP2/overview.aspx). These studies will likely identify additional, unsuspected candidate genes for human disorders.
Unfortunately, these approaches could not be successfully used for most genetic diseases. In these cases, one had to resort to the use of sequence-based strategies to identify the disease gene. As the reader can understand from the previous discussion, this was often a tedious, time-consuming, and expensive process. For rare diseases where the genetic interval defined by genetic linkage was on the order of millions of base pairs, gene discovery was stalled.
Breakthroughs in DNA sequencing technology have revolutionized this process. As indicated in the introduction, there has been an explosion of new high-throughput methods for determining DNA sequence. A comprehensive review of the subject is beyond the scope of this chapter and surely would be outdated by the time this volume is published. Common to all of the methods is the use of massively parallel systems that determine 106 to 108 sequence reads of ˜30 to 400 bp per read in a single experiment. This is in sharp contrast to standard Sanger sequencing machines that maximally determine up to 384 sequence reads of 600 to 1,000 bp per read. Although the new methods, commonly called next generation sequencing, dramatically increase throughput, they also generally have higher error rates and require much greater levels of redundancy to maximize accuracy. Depths of coverage are routinely greater than 20×, and even with this degree of redundancy, gaps and sequence errors can occur. Most laboratories confirm variants of interest with direct Sanger sequencing because of its greater reliability.
There presently are three different approaches for using next generation sequencing for disease gene discovery. In situations where linkage studies have already localized a gene to a chromosome region, investigators use a variety of methods to “capture” the genomic interval and then subject it to next generation sequencing. When linkage is either not possible or when the investigator prefers to use a generally applicable method, he or she pursues either whole genome or whole exome sequencing. As the respective names imply, whole genome sequencing (WGS) determines the sequence of the entire genome, whereas whole exome sequencing (WES, also known as targeted exome capture) restricts its analysis to the DNA sequence of exonic sequences and flanking intron/exon boundaries for all genes. Although WGS is the most comprehensive approach because it includes all regulatory and intronic sequences, its cost is still limiting and the sheer quantity of data it produces presents significant bioinformatic challenges. WES is currently the preferred option because the exome is less than 5% of the size of the entire genome but is estimated to harbor over 85% of all disease-causing mutations.
WES has been successfully used to identify a rapidly growing list of disease genes. In the renal community, this approach was recently used to identify
MY01E (myosin 1E, a podocyte cytoskeletal protein) and
NEIL1 (endonuclease VIII-like 1, a base-excision DNA repair enzyme) as new candidate genes for human autosomal recessive steroid-resistant nephrotic syndrome.
50 In another example, this approach helped to identify
NPHP1 mutations as the cause of disease in two families where consanguinity mapping localized the gene to an interval of almost 30 × 10
6 bp, far too large to tackle with conventional Sanger methods.
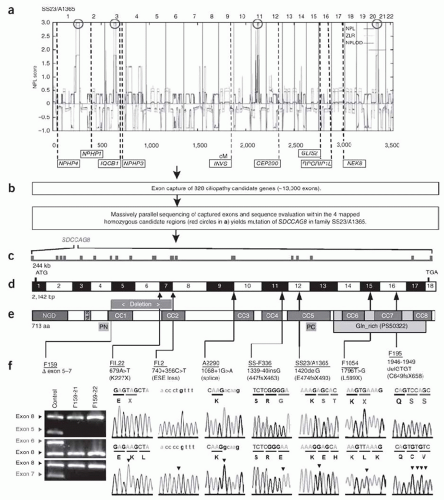
51 Using a variation of this approach, Otto et al.
52 identified mutations in
SDCCAG8 (serologically defined colon cancer antigen 8) as the cause of a nephronophthisis-related ciliopathy (
Fig. 14.1). As the cost drops even further and the technique becomes universally accessible, there is little question that WES is destined to accelerate gene discovery for hundreds of Mendelian disorders.
53In summary, the combination of multiple avenues of biologic data with genetic map position has proved to be a
powerful strategy for finding disease genes. Hence, we have witnessed a remarkable increase in the number of inherited diseases the genetic bases of which have been elucidated.