Introduction
Optimal pharmacological management of renal transplant recipients (RTRs) is imperative when attempting to achieve long-term patient and allograft survival. Short-term outcomes of transplantation have improved considerably over the past 3 decades, due in large part to a more robust understanding of immunology and improvements in surgical techniques, organ procurement, immunosuppression, and post-transplant care [1]. Despite the improvements in short-term survival, late graft loss and complications of long-term immunosuppression remain a concern.
In transplantation, clinical immunosuppression is an empirical practice whose primary goal is to prevent acute rejection while limiting harm to the patient or allograft. Unfortunately, this practice is fraught with imprecision due to the lack of a satisfactory in vivo technique for determining the degree of immunosuppression in patients. Inadequate immunosuppression can result in allograft rejection, whereas overimmunosuppression can lead to significant adverse events, including infection and malignancy. The adverse-event profiles of the individual immunosuppressants necessitate intense post-transplant patient monitoring. Although local protocols and national/international guidelines have been developed to help streamline immunosuppressive strategies, each patient must be managed individually, with constant, careful adjustments of their immunosuppressive and nonimmunosuppressive medications.
Most immunosuppressive protocols are based on the premise that following the transplant, the immune response is at its most vigorous and will require maximal suppression. Over time, host–graft adaptation occurs and less immunosuppression is needed. However, determining the appropriate level of immunosuppression in different patients over time remains difficult. Consequently, most immunosuppressive protocols begin with induction therapy and/or high doses of maintenance immunosuppression in the critical, early post-transplantation period. The doses of maintenance immunosuppression can be steadily reduced over time to a level that the practitioner feels is appropriate for the given patient. In order for this to best be accomplished, it is imperative that clinicians be aware of the specific advantages and disadvantages of the available immunosuppressants, as well as the potential for drug misadventure commonly seen with these agents. This chapter will review the currently available immunosuppressive agents and potential future immunosuppressants, focusing on their pharmacology, dosing, adverse-event profile, drug–drug interaction (DDI), and clinical efficacy.
Immunosuppressive therapies
Most immunosuppressive regimens are built utilizing multiple agents, all working on different immunological targets. The primary reason for a multidrug regimen is to allow for pharmacological activity at several key steps in the T-cell activation/replication process; however, this type of regimen employs lower doses of each individual agent, resulting in less drug-related toxicity.
The three stages of clinical immunosuppression are induction therapy, maintenance therapy, and management of acute rejection. The immunosuppressive therapies used for each of these stages will be discussed in detail in this section. Figure 10.1 depicts the location of action of the current immunosuppressive therapies and immunosuppressants in clinical trials.
Figure 10.1 Locations of action of the current immunosuppressive therapies and immunosuppressants. (Reproduced by permission from John Wiley & Sons Ltd: from [3] Larsen CP, Knechtle SJ, Adams A, Pearson T, and Kirk AD. A Therapeutic Strategy for Long-term Maintenance Immunosuppression. American Journal of Transplantation 2006;6(5):876–883)
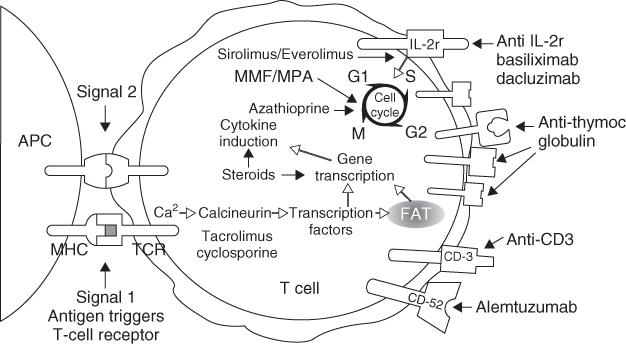
Induction therapy
Induction therapy provides a high level of immunosuppression in the early post-transplant period, when the risk of acute rejection is highest [2]. This stage of immunosuppression is not obligatory; however, given the impact of acute rejection on long-term allograft function, induction therapy is often considered essential to optimum outcome. This is especially true in patients at high immunological risk for rejection. Induction-therapy agents (Table 10.1) are classified as either monoclonal or polyclonal antibodies. Even so, based on their mechanisms of action, these agents may be more clearly delineated by categorizing them as either depleting (OKT3, antithymocyte globulins (ATGs), alemtuzumab) or nondepleting (basiliximab, daclizumab) proteins [2].
Table 10.1 Induction agents used in renal transplant [1,9,28].
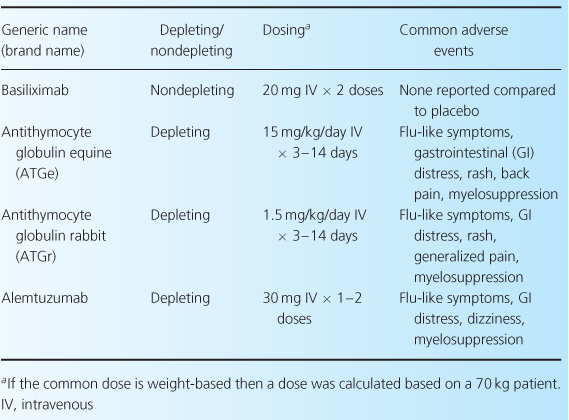
The primary reason for using induction therapy is that these agents are highly immunosuppressive, which allows for a significant reduction in acute rejections rates and improved 1-year graft survival [2,4]. Additionally, these agents are often considered essential for use in patients at high risk for poor short-term outcomes, such as those with preformed antibodies, a history of previous organ transplants, multiple human leukocyte antigen (HLA) mismatches, or who have received organ transplants with prolonged cold-ischemic time or from expanded-criteria donors and donors after cardiac death. Induction therapy can also play an important role in preventing early-onset calcineurin inhibitor (CNI)-induced nephrotoxicity [2,4]. With the use of induction therapy, CNI initiation can often be delayed until the graft regains some function [5].
The short-term improvements in outcomes gained from the use of induction therapies are undeniable, and are discussed in this section, yet studies detailing the impact of induction therapy on long-term allograft function or survival are needed. Practitioners should realize that, when using these highly immunosuppressive agents—particularly the depleting proteins (also known as the antilymphocyte antibodies (ALAs))—host defenses are profoundly impaired, predisposing patients to opportunistic infections and/or malignancies.
Declining worldwide use and manufacturing expenses of both OKT3 and daclizumab have led the makers of these agents to discontinue their production [6]. This review will discuss only those induction agents that remain available.
Nondepleting protein (basiliximab)
Pharmacology
Basiliximab is a chimeric monoclonal antibody that binds with high affinity to and antagonizes the alpha subunit of the interleukin-2 (IL-2) receptor (i.e. CD25) [7,8]. This agent inhibits IL-2-mediated activation of lymphocytes, which is a necessary step in clonal expansion of T-cells [8,9].
Dosing
In adults and children ≥35 kg, intravenous (IV) basiliximab is given at doses of 20 mg 2 hours prior to transplant and again on postoperative day 4 [7,8]. In patients <35 kg, 10 mg doses should be used at the same dosing interval. This dosing strategy saturates CD25 for up to 8 weeks. No dose adjustments are necessary in renal or hepatic dysfunction [7,8].
Adverse events
The incidence of all adverse reactions with basiliximab was similar to placebo in clinical trials [8–10]. On rare occasions (<1% of patients), severe acute hypersensitivity reactions have been reported after both initial exposure and reexposure to this medication. The absence of any increased risk of cytomegalovirus (CMV) infection or malignancy is noteworthy [8–10]. In comparison to induction therapy with ATG, 16 recipients would need therapy with an IL-2-receptor antagonist in order to prevent one case of CMV, and 58 patients would need an IL-2-receptor antagonist to prevent one patient from having malignancy [11].
Depleting proteins (ATGs)
Pharmacology
Polyclonal antibodies directed against human lymphoid tissue are prepared by immunizing animals with human thymocytes [4,7,12,13]. Currently, there are two ATG preparations available in the USA. Antithymocyte globulin equine (eATG) is a purified, concentrated, and sterilized antibody preparation obtained from the hyperimmune serum of horses. This preparation depletes the number of circulating, thymus-dependent lymphocytes through multiple mechanisms. eATG contains antibodies against many T-cell surface markers and promotes T-cell depletion through opsonization with complement-mediated lysis and clearance into the reticuloendothelial system. Immune reconstitution after depletion with eATG takes several months. Overall, the number of lymphocytes in the peripheral circulation, the thymus, and the spleen is reduced [5,7,12,13].
Antithymocyte globulin rabbit (rATG) is also a cytolytic polyclonal antibody preparation [4,7,13]. It is a purified and pasteurized preparation of gamma immunoglobulin derived by immunizing rabbits with human thymocytes. Possible mechanisms by which rATG induces immunosuppression in vivo include T-cell clearance from the circulation and modulation of T-cell activation, homing, and cytotoxic activities. In vitro, rATG mediates T-cell suppressive effects via inhibition of proliferative responses to several mitogens. rATG is thought to induce T-cell depletion and modulation by a variety of methods, including Fc receptor-mediated complement-dependent lysis, opsonization and phagocytosis by macrophages, and immunomodulation leading to long-term depletion via apoptosis and antibody-dependent cell-mediated cytotoxicity. Cell recovery may take several months following rATG administration [4,7,13]. It has been shown that T-cell recovery following rATG-induced depletion is associated with an expansion of suppressor T-cells, potentially inducing long-term immunomodulation [14].
Dosing
The approved induction-therapy dosing strategy for eATG is 10–30 mg/kg/day IV for 7–14 days [5,7,12,13]. The initial dose should be administered shortly before or after transplantation. In many instances, this prolonged course is associated with a difficult outpatient administration and high cost. A shorter course of therapy (15 mg/kg/day for 7–10 days) is often used to counter these issues. rATG is not approved for induction therapy but is routinely used for this purpose. When used in this capacity, rATG has been dosed at 1–4 mg/kg/day for 3–10 days after transplantation. The most common dosing regimen with this agent is four doses of 1.5 mg/kg/day, resulting in a total dosage of 6 mg/kg over the course of therapy [4,7,13]. There is a benefit in giving the initial dose intraoperatively prior to the anastamosis of the renal artery in order to reduce organ reperfusion injury [15].
Adverse events
Cytokine release-related adverse events, such as fever, chills, headache, back pain, nausea, diarrhea, dizziness, malaise, and myelosuppression, are common with both ATG preparations [7]. ATG-treated patients experience significantly more fever, cytokine-release syndrome, infusion-related reactions, and leukopenia compared to those patients receiving an IL-2-receptor antagonist [11]. Premedication with an antihistamine and acetaminophen is recommended to lower the incidence of fever and chills during the infusion. Anaphylactic reactions have occurred with the administration of eATG and the manufacturer strongly recommends the administration of a skin test prior to the first dose of eATG to assess the potential for an allergic reaction. The patient and injection site should be examined every 15–20 minutes for 1 hour after the administration of the test dose. Unfortunately, a negative skin test does not completely rule out the potential for an allergic reaction. The strong recommendation of the skin test makes the use of eATG more difficult than other antibody preparations [5,7,12]. A test dose is not recommended when using rATG.
There is a theoretical concern about retreatment with ATG for management of steroid-resistant acute rejection in a patient who received ATG for induction therapy, due to the potential for increased adverse events and allergic reaction. Although this has never been formally analyzed in a prospective, randomized manner, two retrospective case series suggest that readministration of rATG can be safely carried out in RTRs [16,17].
Comparative efficacy (eATG versus rATG)
Brennan et al. conducted the sentinel study of eATG versus rATG, in which 72 RTRs were randomized to receive rATG 1.5 mg/kg (n = 48) or eATG 15 mg/kg (n = 24) daily for up to 6 days [18]. All patients were maintained on either cyclosporine (CsA), azathioprine (AZA), and prednisone or CsA, mycophenolate mofetil (MMF), and prednisone. At 12 months, patient survival and renal function were similar between the two groups. However, graft survival was higher with rATG (98%) compared to eATG (83%) (p = 0.020) and acute rejection occurred more frequently with eATG (25%) compared to rATG (4.2% (p = 0.014) at 1 year. In terms of adverse events, leukopenia occurred more commonly with rATG (56.3%) than with eATG (4.2%) (p < 0.0001). Despite this, 6-month CMV disease rates were lower in patients treated with rATG (10%) compared to eATG (33%) (p = 0.025). Post-transplant lymphoproliferative disease (PTLD) did not occur in either group [18].
A 10-year follow-up of these patients revealed that event-free survival (48 versus 29%; p = 0.011) and the incidence of acute rejection (11 versus 42%; p = 0.004) were superior with rATG [19]. Mean serum creatinine levels were higher (1.7 ± 0.5 versus 1.2 ± 0.3 mg/dl; p = 0.003) in the eATG group. There were 0.53 quality-adjusted life years gained from rATG induction [19].
Comparative efficacy (basiliximab versus rATG)
Many have theorized that when using depleting proteins, one has to endure a higher incidence of adverse events in order to attain improved efficacy versus a nondepleting protein. Several studies have attempted to prove this by evaluating the differences between basiliximab and rATG; the most referenced one was carried out by Brennan et al. [20]. Per protocol, event-free survival was defined as freedom from a composite endpoint (biopsy-proven acute rejection (BPAR), delayed graft function (DGF), graft loss, and death). Patients were randomized to receive either rATG (1.5 mg/kg/day from day 0 to day 4; n = 141) or basiliximab (20 mg/day on days 0 and 4; n = 137). Maintenance immunosuppression with CsA-modified, MMF, and corticosteroids was given to all patients. At 12 months, the composite endpoint was similar in both groups (rATG = 50.4%, basiliximab = 56.2%; p = 0.34). However, the incidence of BPAR was lower with rATG (15.6%) versus basiliximab (25.5%) (p = 0.02). Myelosuppression occurred more often in the rATG-treated patients. There were more infectious complications seen with rATG compared to basiliximab (85.8 versus 75.2%; p = 0.03). Despite this, CMV infections were significantly more frequent in the basiliximab group (17.5 versus 7.8%; p = 0.02). Five patients in the rATG group and one patient in the basiliximab group developed malignancy [20].
In a recent meta-analysis, when the IL-2-receptor antibodies were analyzed against the ATG preparations it was found that there was no difference in graft loss [11]. Induction with ATG therapy lowered the rates of BPAR at 1 year (eight studies: RR 1.30, 95% CI 1.01–1.67), but with a 75% increase in malignancy (seven studies: RR 0.25, 95% CI 0.07–0.87) and a 32% increase in CMV disease (13 studies: RR 0.68, 95% CI 0.50–0.93). Adverse events, including myelosuppression and cytokine-release syndrome, occurred more often in patients receiving ATG [11].
Depleting proteins (alemtuzumab)
Pharmacology
Alemtuzumab is a recombinant DNA-derived monoclonal antibody directed against the 21–28 kD cell-surface glycoprotein CD52 [7,21]. CD52 is present on nearly all B- and T-lymphocytes, many macrophages, NK cells, and a subpopulation of granulocytes. This agent causes antibody-dependent cell lysis [22]. Lymphocyte depletion with alemtuzumab is rapid and extensive. Lymphocyte counts may take several months to return to pretransplant levels [21].
Dosing
Alemtuzumab is not approved for use in RTRs. There is no consensus on the appropriate dose of alemtuzumab for induction therapy. Early studies used a dose of 20–30 mg IV or subcutaneous on day 0 and again on either day 1 or day 4 post-transplant [22]. However, current studies are evaluating the use of a single 30 mg dose given on postoperative Day 0, theorizing that this dosing regimen will provide similar efficacy, with improved tolerability, to previous dosing strategies [21].
Adverse events
Some common adverse reactions seen with alemtuzumab include anemia, headache, dysthesias, dizziness, nausea, vomiting, diarrhea, myelosuppression, infusion-related reactions, and infection [7]. The use of premedications (e.g. acetaminophen, antihistamines) is advisable, to reduce the incidence of infusion-related reactions [7,21].
One possible concern with alemtuzumab induction is the potential response associated with immune reconstitution. Although data are unavailable in RTRs, a study in multiple sclerosis showed a profound IL-21-driven autoimmune response following alemtuzumab-induced lymphocyte depletion [23]. Further characterization of the immune system following alemtuzumab use in RTRs is needed in order to understand the long-term impact of the use of this agent as induction therapy.
Comparative efficacy
Data exist on alemtuzumab’s efficacy and safety in several observational studies, although only a few randomized controlled trials have been published to date in RTRs. The most cited alemtuzumab analysis was performed by Hanaway et al. [24]. In this prospective analysis, patients were randomized to receive alemtuzumab or either basiliximab or rATG. Patients were stratified according to risk, with 139 high-risk patients receiving alemtuzumab (30 mg × 1 dose; n = 70) or rATG (6 mg/kg total dose; n = 69). In 335 low-risk individuals, alemtuzumab (30 mg × 1 dose; n = 164) or basiliximab (40 mg total dose; n = 171) was administered. At three years post-transplant, BPAR rates in low-risk patients was lower with alemtuzumab compared to basiliximab (10% vs. 22%; p = 0.003). At the same time period, in high-risk individuals, the rates of BPAR were comparable between alemtuzumab and rATG (18% vs. 15%; p = 0.63). Adverse-event rates were comparable among all four treatment groups [24].
Induction therapy: conclusion
Induction therapy with biological agents increased utilization to 82.8% of kidney transplants in the USA in 2009, continuing a 9-year trend [25]. However, there is no universal consensus on the optimal induction agent. The most recent analysis, in 2009, demonstrated that rATG was the most frequently used induction agent in the USA. Depleting antibody therapy was used in 58% of RTRs, about whom information was available. In contrast, the IL-2-receptor antibodies were used in 21.2% of all RTRs [13,25]. Outside of the USA, the UK National Institute for Health and Clinical Excellence (NICE) has recommended the use of basiliximab for induction therapy; however, NICE provides no review or opinion on the use of the ALA agents [26].
Maintenance therapy
The objective of maintenance immunosuppression is to further aid in preventing acute rejection episodes while optimizing long-term patient and allograft survival [1,27]. Immunosuppressants require careful selection and dosage titration to balance the risks of rejection with those of toxicity. The net state of immunosuppression is strongly correlated with the occurrence of some of the most notorious post-transplant adverse events (e.g. infection, malignancy); therefore, it is vital that the degree of immunosuppression be gradually reduced over time [1,27].
During the early years of transplantation there were few choices for maintenance immunosuppression (e.g. AZA, corticosteroids). In the 1980s, the development of CsA revolutionized organ transplantion by reducing rejection rates substantially and improving 1-year graft survival. The evolution of maintenance immunosuppression in the past 2 decades saw an expansion in therapeutic options which further prevent rejection and improve outcomes. The maintenance immunosuppressive agents, in combination with induction therapy, have made it possible to attain acute rejection rates at or below 10% and increase 1-year graft survival above 90%.
There are five distinct classes of maintenance immunosuppressive agent (Table 10.2): CNIs (CsA and TAC), target-of-rapamycin (ToR) inhibitors (sirolimus and everolimus), antiproliferatives (AZA and the mycophenolic acid (MPA) derivatives) co-stimulation blockade (belatacept), and corticosteroids. In general, maintenance immunosuppression is achieved by combining two or more medications from the different classes in order to maximize efficacy and reduce toxicities. Immunosuppressive regimens vary between transplant centers, but most often include a CNI with an adjuvant agent (e.g. ToR inhibitor, antiproliferative), with or without corticosteroids. Selection of appropriate immunosuppressive agents should be patient-specific, and transplant practitioners must take into account the immunosuppressive medication’s pharmacological properties, adverse event profile, and potential for DDIs, as well as the patient’s comorbidities.
Table 10.2 Maintenance immunosuppressive medications [1,9,28].
| Generic name | Common initial dosage | Common adverse effects |
| CsA | 4–5 mg/kg twice a day | Neurotoxicity, gingival hyperplasia, hirsutism, hypertension, hyperlipidemia, glucose intolerance, nephrotoxicity, electrolyte abnormalities |
| TAC | 0.05–0.075 mg/kg twice a day | Neurotoxicity, alopecia, hypertension, hyperlipidemia, glucose intolerance, nephrotoxicity, electrolyte abnormalities |
| AZA | 1.0–2.5 mg/kg once a day | Myelosuppression, gastrointestinal disturbances, pancreatitis |
| MMF | 0.5–1.5 g twice a day | Myelosuppression, gastrointestinal disturbances |
| EC-MPA | 720 mg twice a day | Myelosuppression, gastrointestinal disturbances |
| Sirolimus | 1–10 mg once a day | Hypertriglyceridemia, myelosuppression, mouth sores, hypercholesterolemia, gastrointestinal disturbances, impaired wound-healing, lymphocele, pneumonitis |
| Everolimus | 0.75 mg twice a day | Hypertriglyceridemia, myelosuppression, mouth sores, hypercholesterolemia, gastrointestinal disturbances, impaired wound-healing, lymphocele, pneumonitis |
| Belatacept | Initial Dosing: 10 mg/kg on post-op days 1 and 4, and post-po weeks 2, 4, 6, 8 and 12. Maintenance Dosing: 5 mg/kg starting at the end of week 16 post-transplant and every four weeks thereafter. | Gastrointestinal disturbances, hyper/hypokalemia, headache, peripheral erdema, anemia, leukopenia, hypotension, arthralgia, insomnia. |
| Prednisone | Maintenance: 2.5–20.0 mg once a day | Mood disturbances, psychosis, cataracts, hypertension, fluid retention, peptic ulcers, osteoporosis, muscle weakness, impaired wound-healing, glucose intolerance, weight gain, hyperlipidemia |
CNIs
Pharmacology
The CNIs induce immunosuppression by first complexing with cytoplasmic proteins: CsA with cyclophilin and TAC with FK-binding protein-12 (FKBP-12) [1,7,28]. This complex then binds to and deactivates calcineurin phosphatase, subsequently preventing the dephosphorylation and translocation of nuclear factor of activated T-cells (NFAT). Inhibition of NFAT’s passage through the nuclear membrane reduces the expression of several cytokine genes that promote T-cell activation and expansion, including IL-2, IL-4, interferon-gamma (INF-γ), and tumor necrosis factor-alpha (TNF-α). The end result of inhibiting calcineurin phosphatase is a reduction in cytokine synthesis, with a resultant decline in lymphocyte proliferation [1,7,28].
Pharmaceutics and dosing
CsA is available in two formulations that have very different oral absorption characteristics. The original, oil-based formulation has bile-dependent absorption, resulting in highly variable oral bioavailability. The newer, modified microemulsion formulation has an improved absorption profile that provides a more consistent drug exposure [29]. CsA-modified is the preferred formulation for transplant centers that utilize CsA-based maintenance therapy. The two formulations are not bioequivalent; however, conversion between the two formulations is safe with proper therapeutic drug monitoring (TDM). The labeled initial oral adult dose of CsA following renal transplant is 15 mg/kg/day (range of 8–18 mg/kg/day) administered in two divided doses [1,7,28]. The IV formulation of CsA is used less frequently in renal transplantation due to its high risk of nephrotoxicity. TAC is commercially available in oral and IV formulations. The recommended adult oral dose of TAC following renal transplant is 0.2 mg/kg/day (range of 0.1–0.3 mg/kg/day) administered in two divided doses [1,7,28]. The use of IV TAC carries the risk of anaphylaxis, secondary to its hydrogenated castor-oil component [7]. CNIs should be initiated within 24 hours of transplantation in patients not receiving induction therapy. However, when used in conjunction with induction therapy, dosing may be delayed until some degree of kidney function is seen. The appropriate selection of a starting dose is influenced by the patient’s immunological risk, preexisting disease state, and concomitant immunosuppressants. In rare instances, TAC may be given sublingually if gut absorption is questionable, and concern exists regarding the use of IV TAC [30,31]. A once-daily, extended-release TAC formulation has been approved in some European countries and Canada, but has not yet been approved for use in the USA.
Therapeutic drug monitoring
Monitoring whole-blood CsA and TAC levels is imperative, especially early post-transplantation, given the great interpatient and intrapatient variability of these agents [8,10]. Trough levels (C0) have been the standard monitoring parameter for both CsA and TAC [9,10]. However, for CsA, newer literature has shown that trough concentrations may not have the best correlation with either efficacy or toxicity and suggests that CsA area-under-the-curve (AUC) is best predicted by a 2-hour post-dose (C2) [9–11]. Since C2 levels are drawn shortly after the administration, the serum concentrations are much higher when compared to C0 (800–1500 ng/ml) [9,10]. The long-term benefits of C2 monitoring remain unclear, yet short-term data suggest that it may provide an initial improvement in renal function and reduce the frequency and severity of CsA-induced hypertension. Manufacturer recommendations for CsA and TAC trough levels range from 50 to 400 ng/ml and from 4 to 20 ng/ml, respectively, depending on the degree of the patient’s immunological risk and the time elapsed since the transplant [7]. Many practitioners have argued that the upper limit of trough levels for TAC are too high, as several studies have shown excellent efficacy and safety with levels of less than 12 ng/ml [9]. Target level recommendations are institution- and patient-specific. CNI TDM should not begin until steady state has been reached, which generally occurs 3–4 days after initiation of therapy or dosage change [8–10].
Adverse events
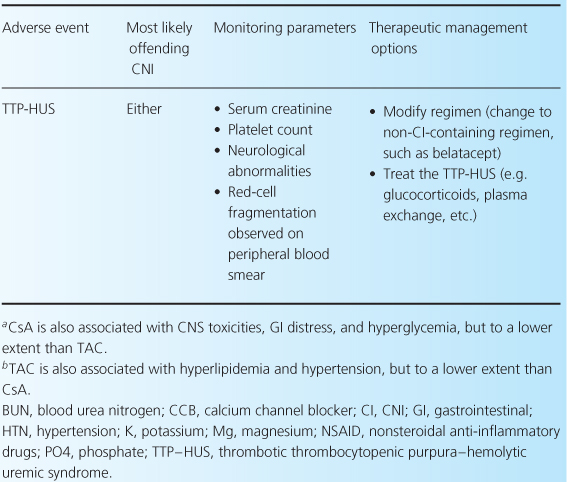
The CNIs have an extensive adverse event profile, with most toxicities being dose-dependent. One of the most infamous effects of both CNIs is their ability to cause nephrotoxicity, as well as other common adverse effects including neurotoxicity, cardiovascular disease, and hyperglycemia [2,4,13]. Rarely, thrombotic thrombocytopenic purpura-hemolytic uremic syndrome can occur while patients are receiving either CNI [32–35]. There are differences between the two agents in terms of adverse-event profiles, which may influence the choice of one agent over the other. For example, post-transplant diabetes, alopecia, and neuropathies are more associated with TAC, while gingival hyperplasia, hirsutism, and hypertension are more commonly associated with CsA. Table 10.3 reviews common CNI adverse events and recommendations for their management.
Table 10.3 Management of common adverse events associated with the CNIs [1,7,28].
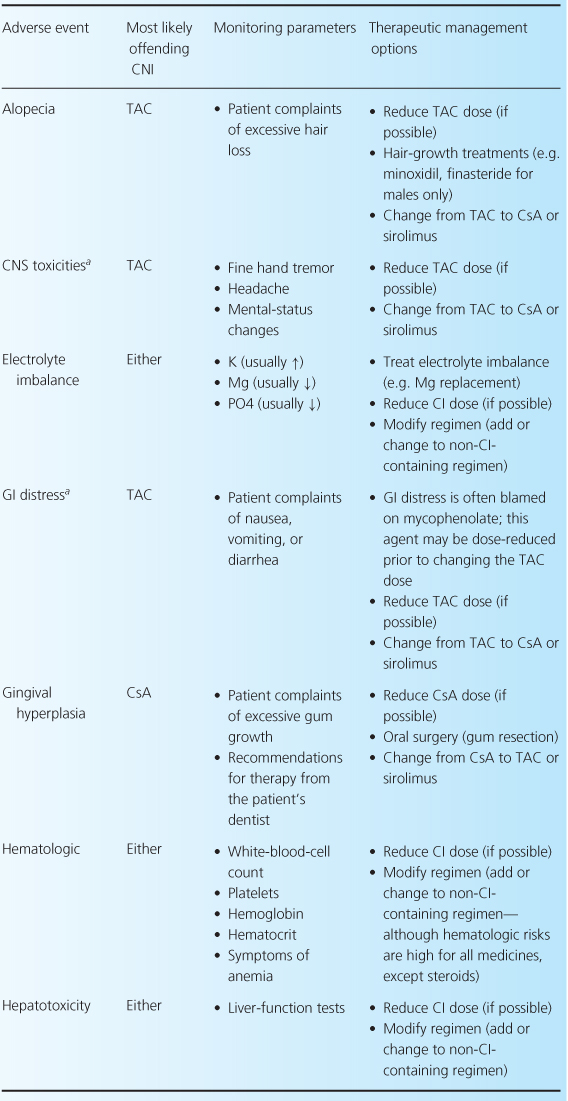
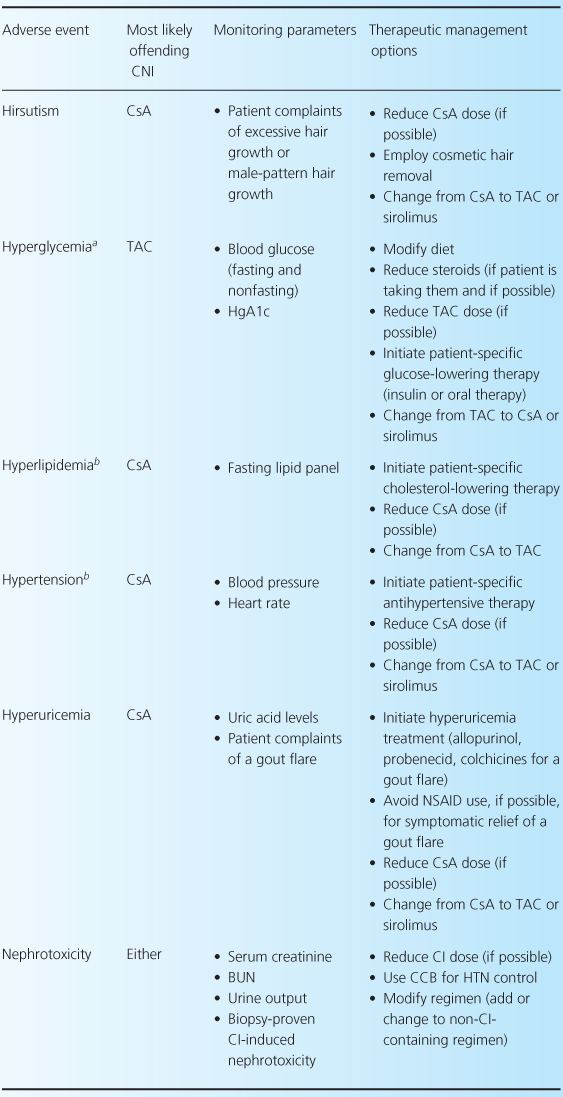
Comparative efficacy
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


