Gastrointestinal diseases
Celiac disease
Crohn’s disease
Ulcerative colitis
Hepatic diseases
Alcoholic liver disease
Hepatic B viral infections
Chronic schistosomiasis
Cryptogenic cirrhosis
Primary biliary cirrhosis
Portosystemic shunts
Autoimmune diseases
Granulomatosis with polyangiitis
Uveitis
Episcleritis/scleritis
Goodpasture’s disease
Chronic thyroiditis
Myasthenia gravis
Vogt–Koyanagi–Harada syndrome
Rheumatological diseases
Ankylosing spondylitis
Reactive arthritis (Reiter syndrome)
Rheumatoid arthritis
Behcet’s syndrome
Sjögren’s syndrome
Systemic lupus erythematosus
Takayasu’s arteritis
Malignancy
Renal cell carcinoma
Bronchial carcinoma
Infections
HIV
Hepatitis A
Hepatitis B
Staphylococcus
Brucellosis
Cytomegalovirus
Dermatological diseases
Dermatitis herpetiformis
Psoriasis
Kidney disease
Nephrotic syndrome with minimal change disease
Membranous nephropathy
Hematologic diseases
IgA monoclonal gammopathy
Mycosis fungoides
Sézary syndrome
Pulmonary diseases
Sarcoidosis
Pulmonary hemosiderosis
Genetics
X-linked thrombocytopenia
Wiskott–Aldrich syndrome
Cystic fibrosis
Renal manifestations may occur before or during the clinical course of the associated diseases, and a wide variety of glomerular lesions may be present. A heavy proteinuria in IgAN is usually indicative of advanced disease, while a true nephrotic syndrome can be due to the combination of IgAN with a “minimal change disease like” podocytopathy or a membranous nephropathy.
Epidemiology
For over 40 years, IgAN has been recognized as a major cause of progressive glomerular disease. In 1987 IgAN was defined as the “commonest glomerulonephritis in the world” [7]. However, its prevalence varies considerably among countries, and it is influenced by sex and ethnicity. It is found more often in men and distinctly less in blacks [8]. Renal biopsy and Registry data of all primary glomerular diseases show that the prevalence of IgAN ranges from 20 % up to 50 % in Asia (45 % in China; nearly 50 % in Japan) and 20–40 % in Europe (37 % in Italy, 29 % in Romania, 19 % in Spain). Percentages as low as 2–4 % have been reported in some parts of the USA. However, a prevalence of over 35 % has been observed in American Indians in the Southwest and 14 % in young American Caucasians. In Australia, the general estimated prevalence is 12 %, but in Victoria, due to a relatively liberal policy of renal biopsy, the prevalence is higher, and the incidence is 87 cases per million per year [9].
Local indications for renal biopsy and health screening practices had influenced the epidemiologic data. Early studies carried out in 1975 reported rates of less than 5 % [10] in Great Britain, while 12 years later, Propper et al. reported that in Scotland, 37 % of patients affected by isolated hematuria had IgAN [11]. Based on autopsy studies on the general population, asymptomatic deposition of IgA is estimated at an average of about 10 % [12].
The majority of studies show a male predominance of at least 2:1 [13]. Unlike other glomerular diseases, IgAN and HSPN are uncommon in both African-Americans and in Africans [14]. Some authors have attributed this difference to a different structural property of IgA2 according to different allotypes, i.e., A2m(1) and A2m(2). The A2m(2) allotype that predominates in blacks is more resistant to cleavage and could provide a protective role against IgAN. Nevertheless, the IgAN clinical course in blacks does not differ significantly from what is observed in whites [15–18].
Genetic Susceptibility
After the initial diagnosis of IgAN in identical twins in 1978 [19], scattered family studies and a number of studies on sporadic cases strongly suggested a genetic predisposition for at least some patients with IgAN and familial primary IgAN. The true frequency of familial IgAN remains uncertain because no serologic markers are currently available. Out of 269 cases from 48 families of IgAN patients, urinary abnormalities have been detected in 23 % [20]. In a second study, the same group examined a cohort of 110 patients with biopsy-proven IgAN and checked for both urinalysis and estimated renal survival. The 20-year renal survival rate from the apparent onset of the disease was significantly shorter in patients with familial (41 %) than sporadic IgAN (94 %). Furthermore, 15-year renal survival from the renal biopsy was reported to be significantly worse in familial disease: end-stage renal disease (ESRD) was present in 64 % of patients with familial and 8 % with sporadic IgAN [21]. Characterization of IgAN1 gene linked to IgAN in some Italian and American multiplex families has remained elusive [22].
A number of immunologic abnormalities suggesting a common genetic substrate affecting B-lymphocyte function in patients and their relatives have been reported: increased serum IgA, multimeric IgA and IgA rheumatoid factor, increased IgA1 production by peripheral blood mononuclear cells, detectable amounts of IgA–IgG complexes, and increased interleukin-2 and interleukin-4.
As regard to immunogenetics, IgAN was initially reported to be associated with HLA Bw35, but this association has not been confirmed. A benign course of IgAN has been associated with HLA DR4 in Japanese patients [23]. Because of the complex genetic architecture of IgAN, efforts to map disease susceptibility genes have not achieved definite results, and no causative mutations have been identified to date. Linkage analysis studies in families with IgAN have identified multiple loci and risk alleles, indicating that IgAN is likely a polygenic disease. These include a major disease locus (IgAN1) on chromosome 6q22–23 in Caucasian families [24] and a number of potentially significant gene polymorphisms, including uteroglobin [25], TGF beta [26], and atherosclerosis-associated genes [27]. A strong association with the HLA locus on chromosome 6p has been recently identified in a genome-wide analysis of familial and sporadic cases of IgAN in white Europeans [28].
Henoch-Schöenlein purpura (HSP) is the most common form of vasculitis in children (with an incidence of 10–20 cases per 100,000 children/year, <10-year-old in 90 % of cases). HLA B35 has been associated with an increased risk of nephritis. HLA DRB1*01, DRB1*11, and DRB1*14 also seem to increase susceptibility to glomerulonephritis.
Recent studies have focused on polymorphisms of genes encoding proinflammatory adhesion molecules associated with endothelial cell activation, TNF-α[alpha] , IL-1β[beta], IL-8, TGF-β[beta], and VEGF [29]. These studies are often limited by the relatively small cohorts.
HSP is a feature of the genetically determined familial Mediterranean fever (FMF) which is often observed in Israel and Turkey [30]. FMF is characterized by neutrophil activation and migration to the serosal surfaces. Neutrophil infiltration of the small vessels is a key histological feature of HSPN. Observation that blood and urinary leukotriene B4 (LTB4) levels are higher while lipoxin A4 (LXA4) levels are lower in patients with HSPN compared to patients without is consistent with this association [31]. Indeed, recruitment of neutrophils and chemotaxis are activated by LTB4 and inhibited by LXA4.
Pathogenesis
Abnormalities in IgA1 Glycosylation
A galactose deficiency or diminished sialic acid content in the hinge region O-linked glycans of IgA1 has been reported in patients with IgAN and HSPN [32]. These results have been recently confirmed in experiments using mass spectrometry proteomic analysis that allows for accurate molecular weight estimation of the various O-glycans [33].
IgA1 has a unique structure at the hinge region and possesses a variable number of O-linked oligosaccharides composed of N-acetylgalactosamine and galactose, which may be sialylated. Studies on immortalized circulating B cells from patients with IgAN have suggested a decreased β[beta]-1, 3 galactosyltransferase enzyme activity that could result in an impaired galactosylation of the core GalNaC residues [34] and an aberrantly exposed GalNac moiety. The origin of the defects in the enzymatic glycosylation of GalNac is unknown. Both genetic abnormalities in enzyme structure or function and acquired perturbations due to microenvironmental influences on IgA1 maturation might contribute. Incidentally, intercurrent infections could determinate the release of microbial neuraminidase, and the epitopes produced by the conformational changes may mimic those of environmental bacterial or viral glycoproteins.
An alternative emerging explanation for the apparent excess of undergalactosylated IgA1 in the circulation of IgAN patients could be that, differently from the response to systemic antigen challenge, the immune response to mucosal antigens is characterized by mucosally derived, relatively galactose-deficient IgA1 [35].
Consequences of the Underglycosylation
Decreased sialic acid and/or diminished content of galactose makes IgA1 molecule susceptible to auto-aggregation, thus forming molecular species whose properties resemble “immune complexes” in the circulation [36]. Galactose-deficient IgA1 is also the target of an “autoimmune” response carried out both by IgA and IgG autoantibodies, thus forming galactose-deficient-IgA1/IgA1 and galactose-deficient IgA1/IgG immunocomplexes [36]. An alternative concept has been proposed in that the increased plasma levels of galactose-deficient IgA1 would not result of an aberrant B-cell production of IgA1 but due to an “aberrant” distribution in which mucosal IgA1-committed plasma cells are misdirected to systemic sites and secrete “mucosal-type” IgA1 into the circulation [35]. The “right” mucosal-type, underglycosylated, IgA1 molecules could circulate in the “wrong” place concomitantly to the “right” anti-GalNac IgG antibodies at the “wrong” time, i.e., when galactose-deficient IgA1 is in circulation. Galactose-deficient IgA1 has a tendency to bind to a variety of glycoproteins, including constituents of normal glomeruli, leading to “planted” antigens, which promote in situ immune complex formation. Moreover, desialylation enables IgA to activate complement through a C4-independent alternative complement pathway promoting deposition and assembly of the membrane attack complex.
Impaired Removal of IgA1 Containing Immune Material from Circulation
Removal from circulation of I-labeled aggregated IgA or I-labeled IgA1–IgG aggregates is delayed in the IgAN patients [37, 38]. The hepatic Ashwell-Morell receptors, originally named asialoglycoprotein receptors (ASGPRs), represent a major pathway of IgA catabolism. ASGPRs bind to desialylated galactose or N-acetylgalactosamine residues in the hinge region of IgA1 governing the removal from circulation of IgA1 and IgA1-containing complex. Studies on the kinetics of intravenously injected neuraminidase-treated radiolabeled orosomucoid have shown normal liver expression of ASGPRs [39]. Undergalactosylated IgA1 could make the normally expressed ASGPR unable to remove IgA1 aggregates or IgA1-containing complexes from circulation. Aberrantly glycosylated IgA1 has been also thought to form immune complexes too large to enter the space of Disse and reach the ASGPRs [40]. However, ASGPRs are known to be the main route mediating the removal of long-term refrigerated platelets [41] casting doubt that immune complexes size prevents access to ASGPRs.
Potential Role of Intercurrent Infection as Cofactors Triggering the Disease
The likelihood of a genetic predisposition of aberrant IgA is supported by the presence of higher levels of serum galactose-deficient IgA1 in patients with IgAN and in their relatives than in healthy controls. However, relatives with high serum levels of underglycosylated IgA1 are mostly asymptomatic, and serum levels of galactose-deficient IgA1 are not elevated in a significant proportion of IgAN patients. Glomerular mesangial IgA deposits in transplanted kidney from donors with subclinical IgAN can disappear in patients without IgAN. Moreover, mesangial IgA deposition is incidentally encountered in asymptomatic individuals: examination of kidney donors and nonselected autopsy series reveal mesangial IgA deposition without clinical evidence of renal disease in 4–16 % of subjects. Thus, it is likely that additional hits are needed for the development of clinical disease.
Environmental factors could contribute. Some patients with IgAN have heightened mucosal sensitivity to certain food antigens [43].
Factors that promote disease expression include high affinity of IgA1-containing immunocomplexes to the mesangium and decreased clearance of IgA-containing immune reactants. Polymeric IgA1 in patients with IgAN binds to mesangial cells with higher affinity than monomeric IgA1 and induces mesangial cellular proliferation. Galactose-deficient IgA1 has a high negative charge and tends to bind to fibronectin, type IV collagen, and laminin.
Infection may trigger autoimmune responses favoring the appearance in circulation of galactose-deficient IgA1/IgA1 and galactose-deficient IgA1/IgG immunocomplexes [36]. A mechanism of molecular mimicry has also been postulated [32]. The conformation of GalNac, which is exposed in galactose-deficient IgA1, is similar to same bacterial and viral epitopes. This reaction could be broadened through intramolecular and intermolecular epitope “spreading” where the primary response against the dominant initiating epitope extends to other epitopes within the same molecule or among different molecules.
Intercurrent infection could also exacerbate disease through activation of Toll-like receptors (TLRs-4). TLRs have been shown to be involved in the switch from IgM to IgA production in B cells. Expression of TRLs-4, whose activation is associated with bacterial lipopolysaccharides, is enhanced in IgAN [44].
Taken all together, the data supports the idea that the abnormalities of the hinge region O-linked glycans make the abnormal IgA1 molecules inherently “sticky” and promote the formation of IgA1 aggregates or immune complexes which escape the physiologic removal systems. Furthermore, these abnormalities increase mesangial deposition of IgA1-containing immune reactants, which is favored by interactions with transferring receptor, fibronectin, or Fcα[alpha] receptor on mesangial cells. Subsequent events include complement activation via the alternative and mannose-binding lectin pathways and release of cytokines that lead to glomerular hypercellularity, matrix production, podocyte injury, and scarring. A numbers of mediator systems are engaged in these processes, including PDGF and TGF-β[beta]. PDGF is intimately involved in the production of mesangial cell proliferation in IgAN. TGF-β[beta] promotes transcription of extracellular matrix glycoproteins and promotes transdifferentiation of tubular epithelial cells to pro-fibrotic myofibroblasts.
Natural History
The clinical course of IgAN is variable. Studies from around the world have shown that about 10 % (3–25 %) of all patients with IgAN have complete resolution of hematuria and proteinuria (although regression of renal lesions and disappearance of mesangial deposits are rare). The most common clinical course is an indolently slow progression to renal insufficiency which occurs in 40–45 % of patients at 20 years of follow-up, half of whom reaching ESRD (i.e., 20–25 % of the total). The actuarial renal survival at 10 years of 4,153 patients recruited from 23 studies in Europe, Australia, Asia, and North America ranged from 77 % to 94 %, 87 % to 93 %, 74 % to 91 %, and 57 % to 67 %, respectively.
This variability is due to diverse diagnostic approaches since a strategy of early renal biopsy in patients with mild urine abnormalities and normal renal function will increase the proportion of IgAN patients with good prognosis. Performing renal biopsy at a late stage will select patients with advanced disease and poor prognosis.
Stable renal function is generally detected in subjects with mild forms of mesangial proliferation, while deterioration of renal function occurs in patients with advanced histological lesions. IgAN prognosis is also more favorable when observation begins at the time of the first symptoms rather than at the renal biopsy [45].
A multicenter study on IgAN collecting data from three continents showed that the overall 10-year renal survival rate was 77 % with a wide range from 93.3 % in Helsinki to 61.4 % in Toronto. Median slope creatinine clearance ranged between −1.24 ml/min/year in Helsinki and −3.99 ml/min/year in Toronto. Taking into account age, creatinine clearance, proteinuria, and mean arterial pressure, this study suggests that variability is mainly related to “lead-time bias” and to the inclusion of milder cases in centers with apparently good outcomes [46]. Further studies are needed to determine whether geographic differences may also be related to genetic factors, environmental factors, or differences in medical management.
Older age at diagnosis has an adverse influence on outcome, while episodes of macroscopic hematuria do not confer a worse prognosis.
A small proportion of patients present with macroscopic hematuria, acute nephritic syndrome, and rapid progression to ESRD over 2–3 years. The main histological features of these cases are crescents (often focal and segmental).
Arterial hypertension, severe proteinuria, impairment of renal function at presentation, as well as histological evidence of glomerular sclerosis and interstitial fibrosis have been reported as strong predictors of unfavorable outcome [45]. Hyperuricemia, increased body mass index, and hyperlipidemia are independent factors of progression [47].
Berthoux et al. calculated the absolute renal risk (ARR) of dialysis. ARR was obtained by counting a number of risk factors present at diagnosis: hypertension, proteinuria >1 g/day, and severe pathological lesions (overall optical score >8) [50]. The cumulative incidence of death or dialysis at 10 and 20 years in adequately treated patients was 2 % and 4 % for ARR = 0, 2 % and 9 % for ARR = 1, 7 % and 18 % for ARR = 2, and 29 % and 64 % for ARR = 3.
Achieving hypertension control and reducing proteinuria lowered the risk of death or dialysis [50].
ARR scoring at diagnosis proved to be useful in estimating at an early stage the final risk of dialysis/death of IgAN patients.
A more complex score using proportional hazard risk models has been proposed to determine the predictors of ESRD in IgAN [51]. In this study, systolic hypertension, proteinuria, hypoproteinemia, azotemia, and high histological grade at initial renal biopsy were independently associated with the risk of ESRD. Mild hematuria was found to predispose patients to ESRD more than severe hematuria. Using eight clinical and pathological variables, a scoring system was developed to estimate 7-year ESRD risk by ROC curve analysis. Patients were subdivided into five groups according to estimated risk (ER), minimum risk (ER = 0–0.9 %), low risk (ER = 1.0–4.9 %), moderate risk (ER = 5.0–19.9 %), high risk (ER = 20.0–49.9 %), and very high risk (ER = 50.0–100 %). The incidence of ESRD over 7 years was 0.2, 2.4, 12.2, 40.2, and 80.8 %, respectively [51].
HSPN is usually a self-limited condition that lasts an average of 4 weeks. A considerable minority of patients relapse. However, relapses usually subside after 6 months. Nephritis occurs in 40 % of patients (within 1 month from onset in 85 % of cases and in nearly all within 6 months). Persistent purpura, severe abdominal symptoms, and an older age are significant risk factors for later nephropathy. Nephritis is a unique feature with potentially chronic consequences that influences the long-term prognosis. It could be estimated that 2 % of all HSPN patients from unselected series will develop chronic kidney disease, but the risk of progression increases up to 20 % at 20 years in tertiary centers. In general, patients with microscopic hematuria and low-grade proteinuria have an excellent prognosis. By contrast, nephritic or nephrotic syndrome has a poor prognosis, with 20–40 % of patients developing long-term renal impairment. A poor correlation between histological features and ultimate outcome has been reported [29].
Clinical Presentation and Diagnosis
The broad clinical spectrum of IgAN includes isolated urinary abnormalities (IUA), acute nephritic syndrome, nephrotic syndrome, acute renal failure (ARF), chronic renal failure (CRF), and rapidly progressive renal failure (RPRF).
Approximately 40–50 % of patients with IgAN present with one or more recurrent episodes of macroscopic hematuria, beginning 1–3 days after an upper respiratory tract infection (synfaringitic hematuria). The episode usually ends after a few hours or a few days, although microscopic hematuria may persist indefinitely. During the episodes the patient usually presents low-grade fever, loin pain (stretching of the renal capsule) mimicking urinary tract infection, or urolithiasis [8, 52–54]. Less commonly, patients may present with acute renal failure. Patients in whom acute renal failure is associated with macroscopic hematuria (ARF-MH) show a 75 % rate of complete recovery [55]. The most striking histological abnormalities in ARF-MH are signs of acute tubular necrosis with a high proportion of tubules that are filled with red cells casts.
The risk factors for incomplete recovery of renal function after the disappearance of MH include MH lasting longer than 10 days, patient’s age >50 years, decreased baseline estimate glomerular filtration rate (eGFR), absence of previous episodes of MH, and severity of tubular necrosis [55]. When IgAN presents with a rapidly progressive glomerulonephritis, the histological pattern is usually a crescentic GN. Most of these patients progress to ESRD within 2–3 years [3, 56, 57].
Thirty percent of IgAN patients show an indolent course characterized by microscopic hematuria and mild proteinuria [58] incidentally detected on routine examination [59, 60]. Gross hematuria occurs in 20–25 % patient.
Patients with persistent microscopic hematuria without proteinuria, normal renal function, and no episodes of macroscopic hematuria will have a diagnosis of thin basement membrane nephropathy (TBMN) or variants of Alport’s syndrome in 43 % of cases and IgAN in 20 % [59]. Conversely, patients with microscopic hematuria associated with low grade of proteinuria will have a diagnosis of IgAN in 46 %, TBMN in 7 %, and other nephropathies in 26 % of cases.
Routine renal biopsy is indicated for asymptomatic microscopic hematuria with low-grade proteinuria [59], and some countries have started routine screening for urinary abnormalities among the general population and promoting a policy of early renal biopsy [12].
In children, IgAN is the second cause of nephritic syndrome after postinfectious glomerulonephritis [61].
Nephrotic syndrome associated with mild mesangial proliferation and diffuse fusion of the foot processes at electron microscopy may result from two simultaneous diseases, i.e., minimal change disease plus a mild form of IgAN [62]. This hypothesis is supported by a rapid remission of proteinuria induced by corticosteroids [63].
By contrast, Kim et al. [64] examined clinical features and long-term outcome of more than 1,000 biopsy-proven IgAN patients. In almost a 100 patients, nephrotic syndrome occurred in any subclass of IgAN, and corticosteroids administration did not result in complete reduction of heavy proteinuria. These findings suggest that MCD is not entirely responsible for the development of NS in patients with IgAN. Complete remission (CR), partial remission (PR), and no response (NR) occurred in 48 %, 32 %, and 20 % of patients, respectively. The prognosis of NS in IgAN was not favorable unless PR or CR was achieved. The risk of doubling the baseline serum creatinine was significantly higher in the NS group than in the non-NS group (p < 0.001) [64].
Overlapping IgAN and membranous disease have rarely been described [65].
In a few cases, IgAN was described in patients with granulomatosis and polyangiitis [66].
In an adult male with gross hematuria, especially if he is a smoker or complains of loin pain and low-grade fever, a urological cause of macroscopic hematuria should be ruled out: kidney, bladder or prostate cancer, stones, or urinary tract infections including tuberculosis. In these patients, a CT urogram and cystoscopy should be considered. If the urological study is negative and the patient shows persistent prevalently dysmorphic microscopic hematuria and mild proteinuria, renal biopsy is recommended. Elevated serum IgA levels as well as elevated serum IgA/C3 concentration ratios (>4.0–4.5) can be seen in about 50 % of patients. Serum IgA/C3 concentration ratio >3, in the presence of overt proteinuria and elevated blood pressure, is said to distinguish IgAN from other causes of hematuria in over 75 % of cases [67].
Circulating IgA rheumatoid factor and IgA immunocomplexes, IgA–fibronectin complexes, IgA deposits in the dermal capillaries at skin biopsy, elevation of plasma polymeric IgA1 levels, measurement of poorly galactosylated IgA1 o-glycoforms in the serum, and urinary proteomics have all been proposed as diagnostic biomarkers. None of them have proven to be useful in clinical practice [68–73]. Serum complement levels (C3, C4, and C1q) are normal and C-reactive protein (CRP) levels are not increased [74]. In some patients, positive ANCA test (IgA subclass) has been reported using purified antigens [75].
Pathological Features
Light Microscopy
IgAN can show different patterns of glomerular injury, although the mesangial area is predominantly involved [76]. The clinical syndrome is often suggestive of a specific histological lesion [1]. Hematuria, mild proteinuria, and normal renal function are often related to mild mesangial proliferation. Moderate to heavy proteinuria and/or acute renal impairment is associated with varying degrees of either endocapillary or extracapillary proliferation, while chronic renal impairment is often observed when glomerular sclerosis is present.
The diverse prevalence of minimal glomerular changes in worldwide studies possibly depends on the biopsy policy [77].
Glomeruli
. All forms of glomerular injury have been described in IgAN. In most cases, the disease appears as a mesangial proliferative GN with focal (less than 50 % of glomeruli involved) (Fig. 8.1) or diffuse (more than 50 %) mesangial hypercellularity (defined as more than three mesangial cells per mesangial area), which is almost always accompanied by reactive mesangial matrix expansion. If patency of the capillary lumens is compromised by the expanded matrix, it is defined as segmental or diffuse sclerosis.
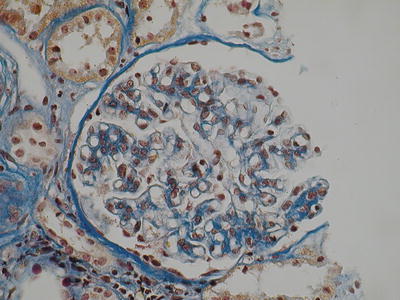
Fig. 8.1
Mild mesangial hypercellularity (AFOG, ×400 magnification) (Class. Oxford: M1, S0, E0, T0)
Another pathological pattern is focal or diffuse endocapillary proliferative GN (Figs. 8.2 and 8.3). This differs from mesangial proliferative GN by the presence of nonresident cell infiltrates (lymphocytes, monocytes, or granulocytes, according to the phase of the inflammatory process) that may result in narrowing of the capillary lumen. Occasionally, subendothelial deposits can be seen and are characterized by nonresident and mesangial cell infiltration of the capillary wall. Glomerular hyperlobulation and “double contour” membranoproliferative appearance can be rarely observed.
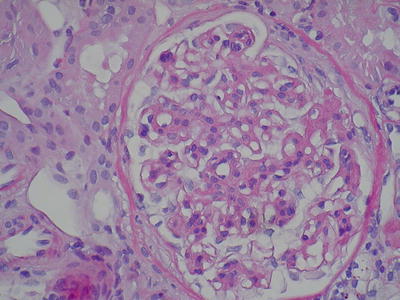
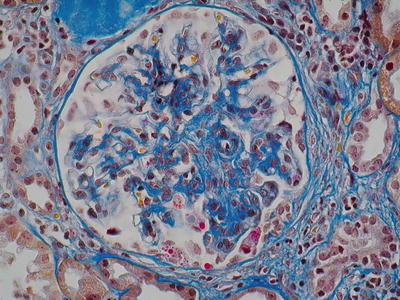
Fig. 8.2
A glomerulus with diffuse mesangial hypercellularity and mesangial matrix with a focal membranoproliferative pattern. Segmental adhesion to the Bowman capsule (H&E, ×400) (Class. Oxford: M1, S1, E1, T1)
Fig. 8.3
Mesangial expansion and focal endocapillary proliferation (AFOG, ×400) (Class. Oxford: M1, S0/1, E1)
In some biopsies, huge deposits of IgA/IgG can be identified by LM stained by fuchsin and PAS. Segmental necrosis of the loop and fibrin deposition, which evolves into adhesions or crescents, can be also observed (Fig. 8.4). Extracapillary GN can be found in 25 % of IgAN cases. An ANCA-associated vasculitis [78–80] should be ruled out in these cases.
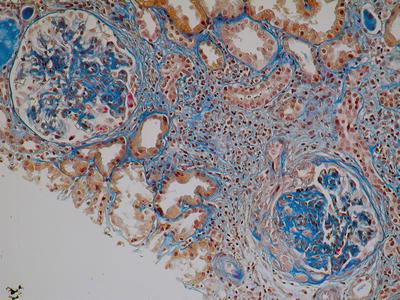
Fig. 8.4
Mesangial hypercellularity with expanded matrix and segmental fibrosis. In the lower right glomerulus, a segmental cellular crescent with adhesion and fragmentation of Bowman capsule. There is also an interstitial fibrosis with inflammatory infiltration associated with tubular atrophy (AFOG, ×400) (Class. Oxford: M1, S1, E1, T1)
Severe inflammation, especially if untreated, results in focal or diffuse glomerulosclerosis.
The heterogeneity of IgAN makes it possible to find nonspecific lesions with negative IgA fluorescence in early biopsies and clear-cut features in subsequent biopsy specimens (i.e., lead-time bias).
Tubuli and Interstitium.
Tubuli and interstitium are involved mainly in chronic stages. Tubulointerstitial injury is on occasion characterized by eosinophilic cell infiltration as well as mononuclear and plasma cell infiltration, all of which may lead to chronic interstitial fibrosis (Fig. 8.4). In cases of macro- and less frequently microscopic hematuria, stacked casts of dysmorphic red blood cells can be seen within the tubules. Occasionally, massive hematuria and acute renal tubular damage can lead to an increase in serum creatinine. Persistent micro-/macro-hematuria is suggested by the presence of hemosiderin in the interstitium, and especially in the medulla.
Vessels.
Vessels are usually normal in the early stages of disease, but vascular sclerosis may be present in more advanced cases.
Immunofluorescence
On immunofluorescence IgAN is characterized by diffuse IgA mesangial immune deposits (Fig. 8.5) which are dominant or codominant compared to concurrent IgG and IgM deposits [81]. Deposits can be also segmentally distributed along the glomerular basement membrane. IgG deposits are found in up to 85 % of the cases. IgM is detectable in sclerotic areas, mainly because of entrapment phenomena. C3 complement fraction is a common finding (90 %). Occasionally, C4d (30 %) and C1q (10 %) can overlap with IgG or IgM deposits, suggesting activation of the lectin complement pathway [82–84]. There is usually a prevalence of lambda chain [85].
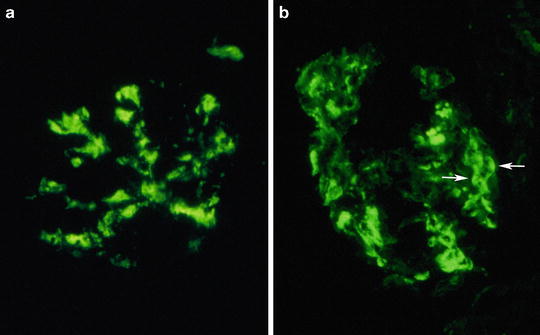
Fig. 8.5
Deposition of IgA immunoglobulin in the mesangium (a) and within mesangium and along capillary membrane (b). Paramesangial prominent deposition (arrows) can form images resembling arc, named “arc de cercle” by some authors (original magnification ×400)
Fibrinogen and fibrin are found in extracapillary forms.
Electron Microscopy
Ultrastructural analysis can be important to define the nature and the site of the deposits. The deposits can be located in mesangium and para-mesangium (100 %) (Fig. 8.6) in subendothelial (11 %) or subepithelial areas (6 %). Rarely, intramembranous (2 %) and hump-like deposits can also be seen.An association between IgAN and TBGM has been described, with TBGM present in 5 % of patients with IgAN [86].
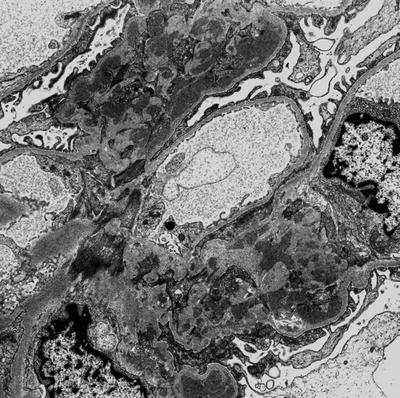
Fig. 8.6
At the electron microscopy glomerular IgA deposition appears as fine granular electron dense material localized within mesangium, with marked accumulation along paramesangial basal membrane (arrows) (original magnification ×3,000)
Prognostic Impact of Histological Classification
Glomerulosclerosis, tubular atrophy, and tubular fibrosis as well as mesangial and endocapillary proliferation and extensive crescents are histological predictors of bad prognosis. A number of classifications have been proposed to identify prognostic factors in IgAN [3, 87–89].
Recently, the Renal Pathology Society together with the International Group of IgAN Network supported an international consensus on the predictive role of the main histological lesions, the Oxford classification [90, 91]. Two hundred and sixty-five biopsies from patients with IgAN with proteinuria greater than 0.5 g/24 h and eGFR more than 30 ml/min/1.73 m2 at the time of diagnosis who had been followed up for at least 1 year were included. Twenty histological lesions were identified, and six of particular relevance were selected on the basis of frequency and reproducibility: mesangial proliferation and endocapillary proliferation, crescents, segmental sclerosis and synechiae, tubular atrophy and interstitial fibrosis, and arterial score. Four of these variables, (1) the Mesangial hypercellularity score, (2) Segmental glomerulosclerosis, (3) Endocapillary hypercellularity, and (4) Tubular atrophy/interstitial fibrosis, were able to predicting renal outcome independent independently of clinical assessment (Table 8.2). Validation of the Oxford classification has been carried out in children as well as in adults [92–94]. Verification is necessary because premature implementation of biased conclusions can harm patients and mislead future research. In some studies mesangial hypercellularity and segmental glomerulosclerosis were weaker predictors than others [93, 94]. In a retrospective analysis of 187 adults and children with IgAN, the four individual variable of MEST offered the same predictive value in both cohorts except mesangial hypercellularity, which was a weaker predictor than in the original Oxford classification paper [94]. Bellur et al. compared the immunofluorescence pattern with optical findings categorized according to the Oxford classification score system and demonstrated the supportive role of IgG and the deposition of IgA in the capillary wall in predicting the development of proliferative glomerulopathy [95]. The Oxford classification has also been validated for in children with IgAN [93, 96]. Recently, Alamartine et al. carried out a validation in 183 adult patients who had IgAN and were followed for an average of 77 months. In univariate time-independent analyses T, E, and S lesions of MEST score were strongly associated with doubling creatinine or ESRD. Conversely, M lesion was not associated with renal outcome. In the multivariate model, only estimated GFR at baseline was a risk factor, and pathological lesions had no independent influence [97]. Proteinuria and blood pressure also failed to predict survival independently from a combined event when factoring in the initial eGFR.
Table 8.2
Past and present IgAN classifications
• Lee histological classification (1982) |
⚬ I: Focal mesangial expansion |
⚬ II: Moderate focal proliferative |
⚬ III: Mild diffuse proliferative |
⚬ IV: Moderate diffuse proliferative; crescents in ≤45 % of glomeruli |
⚬ V: Severe diffuse proliferative; crescents in >45 % of glomeruli |
• Haas histological classification (1997) |
⚬ I: Minimal or no mesangial hypercellularity |
⚬ II: Focal and segmental glomerulosclerosis without active cellular proliferation |
⚬ III: Focal proliferative glomerulonephritis |
⚬ IV: Diffuse proliferative glomerulonephritis |
⚬ V: ≥40 % globally sclerotic glomeruli or area of cortical tubular atrophy |
• Four key pathological features of the Oxford classification (2009)—MEST |
⚬ Mesangial hypercellularity |
(a) Mean < 4 mesangial cells/mesangial area (M0 score) |
(b) Mean ≤ 4 mesangial cells/mesangial area (M1 score) |
⚬ Endocapillary hypercellularity |
(a) Present (E0 score) |
(b) Absent (E1 score) |
⚬ Segmental glomerulosclerosis or adhesions |
(a) Present (S1 score) |
(b) Absent (S0 score) |
⚬ Tubular atrophy/interstitial fibrosis |
(a) 0–25 % (T0 score) |
(b) 26–50 % (T1 score) |
(c) >50 % (T2 score) |
Validation studies include categories of patients that are similar but not identical to the original study so that duplicating every result is not expected [98]. Despite the rigor of the methodology adopted, the Oxford classification remains the result of a retrospective observation, and although special attention was paid to search for independent risk factors, confounding factors cannot be ruled out. Prospective studies are therefore needed to further evaluate these results. For instance, since cases who had been followed up for less than 1 year have been not examined, a number of patients with rapidly progressive IgAN or advanced CKD have been excluded. Conversely, excluding patients with proteinuria <0.5 g/24 h means excluding cases with more favorable prognosis. The involvement of patients from eight different countries made the cohort more varied and representative, though it was not possible to guarantee the homogeneity of the laboratory findings. Analyzing renal survival using two different statistical methods of multivariate analysis provided different results for segmental glomerulosclerosis and for mesangial hypercellularity. The histological study was carried out with PAS staining alone, which, by itself, is not ideal for all the lesions observed in the first stage of the study. Besides, no immunofluorescence or electron microscopy correlations have been investigated. These issues could be the aims of further refinement of the classification.
Treatment
As previously discussed, IgAN is characterized by a variable clinical profile (ranging from a benign course to rapidly progressive renal failure) and a wide spectrum of histological lesions.
Up to 23 % of patients, i.e., those who do not develop proteinuria >300 mg/24 h or hypertension during the follow-up, will have complete clinical remission [99]. Patients whose urinary protein excretion increases during the follow-up are more likely to develop renal impairment compared to patients without proteinuria [99].
Proteinuria is the most important predictor of the decline of GFR rate in studies using multivariate analysis [100]. Five patterns of progression have been identified on the degree of proteinuria: <0.3 g/day, 0.3–1 g/day, 1–2 g/day, 2–3 g/day, and >3 g/day. The kidney survival curve at 15 years was 96 %, 87 %, 65 %, 55 %, and 30 %, respectively. Nevertheless, patients presenting with baseline proteinuria >3 g/day who achieved partial remission, especially if proteinuria dropped to levels less than 1 g/day, had a course similar to patients presenting with <1 g/day at diagnosis [100]. A treatment goal of <0.5 g/day per 1.73 m2 is recommended in children [101]. Values of proteinuria equal or less than 0.5 g/day [102] have been reported as a threshold of risk in adults as well.
Uncontrolled hypertension is associated with greater proteinuria and faster decline of GFR [89, 103], while reducing blood pressure has protective effects especially in mild renal insufficiency [103]. In order to achieve maximal renal and cardiovascular protection, blood pressure goals <130/80 mmHg in patients with less than 300 mg/24 h of proteinuria, and <125/75 in patients with proteinuria >1 g/24 h are recommended [104–106].
Low GFR at presentation was also listed among the clinical factors of poor prognosis [45], but this issue remains controversial [89, 107].
KDIGO guidelines recommend the assessment of all patients with biopsy-proven IgAN for secondary causes of disease, emphases on the role of pathological findings in assessing prognosis, and the evaluation of the risk factors of progression (i.e., proteinuria, blood pressure, and eGFR) at the time of diagnosis and during the follow-up [108–113].
Conservative Treatment
Conservative treatment is based on hypertension control with a low salt diet, the use of angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers, and the use of statins. Tonsillectomy and fish oil have been also proposed for patients with IgAN.
Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers
Angiotensin-converting enzyme inhibitors (ACEi) and angiotensin II receptor blockers (ARBs) reduce the risk of progressive renal disease in a number of proteinuric diabetic and nondiabetic glomerulopathies [114]. One observational study [115] and three small prospective trials [102, 116, 117] have confirmed initial observations [118, 119] that ACEi protect long-term renal function in patients with severe IgAN and reduce proteinuria in normotensive IgAN patients. Four more recent studies provided evidence that ACEi and ARBs are more effective than other antihypertensive drugs in reducing proteinuria and slowing the progressive decline in GFR in IgAN. A 3-year prospective follow-up study showed that the decline in creatinine clearance was lower in ACEi-treated group as compared to control patients treated with amlodipine [115]. Positive effects of reducing proteinuria in slowing the progression of renal damage can be seen with proteinuria values greater than 500 mg/day. In these cases ACE inhibition should be given even in normotensive patients.
Fish Oil
The rationale for using n-3-polyunsaturated fatty acids (omega-3 fatty acids) is the potential anti-inflammatory effect and reduction of glomerulosclerosis. Donadio et al. studied 106 patients with a mean baseline creatinine clearance of 82 ml/min and protein excretion of 2.5–3.2 g/day who were randomly assigned to therapy with either 12 g of fish oil or a similar amount of olive oil (placebo) for 2 years [122]. Baseline clinical characteristics were similar between the two groups. At 4 years, 6 % of the patients in the fish oil-treated group vs. 33 % of placebo group showed a >50 % increase in serum creatinine. ESRD occurred in 10 % of the patients in the fish oil-treated groups vs. 40 % of placebo groups and 15 % vs. 37 % at 6 years of follow-up. In a trial of Southwest Pediatric Nephrology Study Group [123] 96 patients were randomly assigned to one of three treatment arms: a purified preparation of omega-3 fatty acids (4 g/day) for 2 years, alternate-day prednisone (60 mg/m2 per dose for 3 months, 40 mg/m2 per dose for 9 months, and 30 mg/m2 per dose for 1 year), or placebo. The superiority of omega-3 fatty acids over placebo in slowing progression of renal disease was not confirmed at 3 years. A Cochrane meta-analysis of four trials showed no beneficial effect of fish oil on renal outcomes including serum creatinine, creatinine clearance, or change in proteinuria [124]. However, fish oil supplements have shown a number of cardiovascular beneficial effects: lowering of systolic blood pressure and triglyceride values, reduction of resting heart rate, improvement in several markers of endothelial damage, and reduction in the risk of sudden cardiac death in patients with established coronary heart disease. KDIGO suggests the use of fish oil in patients with IgAN and persistent proteinuria > 1 g/day [108].
Lipid Control
In a meta-analysis that included 13 prospective controlled trials in 404 patients, mostly diabetics, Fried et al. examined the effects of lipid-lowering agents on GFR and proteinuria or albuminuria in patients with renal disease [125]. The pooled outcome showed only a trend towards benefit by lipid-lowering therapy (mainly, but not exclusively, statins) on protein or albumin excretion. Several other studies have failed to confirm any effect. Long-term statin treatment failed to reduce proteinuria in 16 patients with NS and renal insufficiency [126]. No effects in children with steroid-resistant NS or in nephrotic patients with MN have been observed [127–129]. Taken all together, the anti-proteinuric effect of statins appears to be negligible. Statins are perhaps more effective when used in combination with ACEi, although the majority of the data has been obtained from patients without NS. On the other hand, proteinuria has been reported as a complication of statin therapy [130–132] as a consequence of statin inhibition of protein uptake by tubular cells [133, 134] which could be dose related.
Related posts:
 General Approach to the Diagnosis and Management of Glomerular Diseases
Childhood Onset Nephrotic Syndrome
General Approach to the Diagnosis and Management of Glomerular Diseases
Childhood Onset Nephrotic Syndrome
 Membranoproliferative Glomerulonephritis
Membranoproliferative Glomerulonephritis
 Thrombotic Microangiopathies: Thrombus Formation Due to Common or Related Mechanisms?
Thrombotic Microangiopathies: Thrombus Formation Due to Common or Related Mechanisms?
 Glomerular Disease in Pregnancy
Glomerular Disease in Pregnancy
 Anti-glomerular Basement Membrane Disease
Anti-glomerular Basement Membrane Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


