Joel B. Nelson, MD
Historic Overview
The response of prostate cancer to androgen ablation is among the most reproducible, durable, and profound of any systemic therapy for a solid tumor. The early and frequent descriptions of immediate relief of bone pain from metastatic prostate cancer after castration do not diminish the marvel of observing this phenomenon first hand. As is the case with many paradigm-shifting observations, endocrine therapy was based on a simple hypothesis. Described as a “biological syllogism,” the idea had a major premise—“In many instances a malignant prostatic tumor is an overgrowth of adult epithelial cells,” a minor premise—“All known types of adult prostatic epithelium undergo atrophy when androgen hormones are greatly reduced in amount,” and a conclusion—“Therefore, significant improvements should occur in the clinical condition of patients with far advanced prostate cancer subjected to castration” (Huggins, 1947).
It had been known for at least a century that prostatic epithelium undergoes atrophy after castration (Hunter, 1840). The breakthough in Huggin’s hypothesis was the recognition that benign prostatic epithelium and prostate carcinoma were biochemically analogous and they would respond in a similar fashion to androgen ablation. Emphasizing the importance of basic observations—“The evidence for the facts which represent the premises was obtained entirely in the laboratory” (Huggins, 1944)—studies on acid phosphatase provided the crucial link between benign and malignant prostate cells. Large amounts of acid phosphatase were found in the prostate glands of men and monkeys (Kutscher and Benjamin, 1935) and in primary and metastatic prostate cancer (Gutman et al, 1936), and the levels increased with androgen administration (Gutman and Gutman, 1938a). Serum levels of acid phosphatase were increased in men with disseminated prostate cancer (Barringer and Woodard, 1938; Gutman and Gutuman, 1938b). With localization of the enzyme to prostatic epithelial cells and primary and metastatic prostatic cancer cells (Gomori, 1939), the stage was set for Charles Huggins, R. E. Stevens, and Clarence V. Hodges to test the hypothesis in men with prostate cancer.
Despite negative results of castration in two men with prostate cancer reported by Young (1936), a series of 21 consecutive patients with locally advanced or metastatic prostate cancer underwent surgical castration at the University of Chicago. “A noticeable improvement occurred in the clinical status of all but three patients” with weight gain, resolution of anemia, and improvement in pain (Huggins et al, 1941). Other reported consequences of castration—a large appetite for food, loss of sexual desire and penile erections, and hot flashes—remain the common side effect profile of androgen ablation therapy today. Although this report was the first to describe the benefits of androgen ablation in the treatment of prostate cancer it also created a new disease state: androgen-refractory prostate cancer.
In considering these “failure cases” (Huggins, 1942), it was found that those with small testes at the time of castration had a poor prognosis, the first description of a more ominous prostate cancer arising in the hypogonadal man. After castration, rises in the levels of urinary 17-ketosteroids, a major metabolite of the adrenal gland, led to the hypothesis that adrenal androgens contributed to subsequent disease progression. The first reports of bilateral adrenalectomies for the treatment of castration-resistant disease (Huggins and Scott, 1945) are described later in a somewhat defensive manner (Scott, 1954), owing, perhaps, to the lack of response and high perioperative mortality. Hypophysectomy and pituitary irradiation (Murphy and Schwippert, 1951) were also investigated. Unfortunately the benefits of surgical castration were soon equaled by the tenacity and inevitable progression of androgen independence, a state still synonymous with the lethal form of the disease. Even in accepting the Nobel Prize for this work in 1966, Charles Huggins admitted “Despite regressions of great magnitude, it is obvious that there are many failures of endocrine therapy to control the disease” (Huggins, 1972).
Direct ablation of the source of androgen, like surgical castration, is only one of the perturbations of the hypothalamic-pituitary-gonadal axis developed to treat prostate cancer. The first central inhibition of the axis exploited the potent negative feedback of estrogen on luteinizing hormone (LH) secretion: it is now known that estradiol is a 1000-fold more potent at suppressing LH and follicle-stimulating hormone (FSH) secretion by the pituitary compared with testosterone (Swerdloff and Walsh, 1973). The effects of estrogen on the male phenotype, namely, regression of androgen-sensitive tissues, have been exploited, historically, to produce the effects of castration without surgical removal of the testes. For example, capons (neutered roosters) were produced by placing estrogen pellets in the neck of the bird rather than by castration (Scott, 1954). Among the various estrogenic compounds diethylstilbestrol (DES) has been most widely studied and used. Early studies indicating improved survival in men treated with both surgical castration and continuous DES (Nesbit and Baum, 1951) have not held up under further scrutiny, but the equivalence of DES compared with castration has. Indeed, given the effectiveness of the considerably less expensive estrogenic compounds it is unfortunate the associated cardiovascular toxicity has limited their widespread use.
The first isolation of luteinizing hormone–releasing hormone (LH-RH) by Schally and colleagues (1971, 1992) required the hypothalami of 165,000 pigs to obtain 800 µg of the 10 amino acid peptide. This 1977 Nobel Prize–winning work led to the development of synthetic LH-RH analogues, peptides generated by substituting D-amino acid residues at certain locations in the natural compound, creating both LH-RH agonists and LH-RH antagonists. After an initial surge in LH release (and testosterone levels) in response to LH-RH agonists the loss of phasic pituitary stimulation results in plummeting LH levels. In the absence of LH, Leydig cell production of testosterone drops to castrate levels. Initially, the clinical utility of these agents was hampered by their short half-lives, requiring daily injections to maintain suppression of the hypothalamic-pituitary axis. The generation of long-acting depot preparations, lasting several months, has established LH-RH agonists as the dominant treatment in hormone therapy for prostate cancer. Recently, direct LH-RH antagonists have been developed for clinical use. Lacking agonist action, these agents do produce the surge in LH and testosterone. It is interesting that both classes of compounds were developed within a few years of the discovery of LH-RH and yet it took decades to develop clinically useful agents.
Molecular Biology of Androgen Axis
The androgen receptor (AR) is a member of the nuclear receptor superfamily, which includes the sex steroids (androgen, estrogen, progestin), adrenal steroids (mineralocorticoids, glucocorticoids), thyroid hormones, vitamin D, and retinoids. These receptors act as ligand-inducible transcription factors, meaning that they cause transcription of target genes within specific cells after ligands (e.g., testosterone) bind to them. All current forms of androgen-deprivation therapy (ADT) function by reducing the ability of androgen to activate the AR, whether through lowering levels of androgen or by blocking AR binding (Fig. 109–1). Therefore the AR is not directly affected by ADT, leading many to hypothesize that castration-resistant prostate cancer is a result of reactivation of AR-mediated pathways.
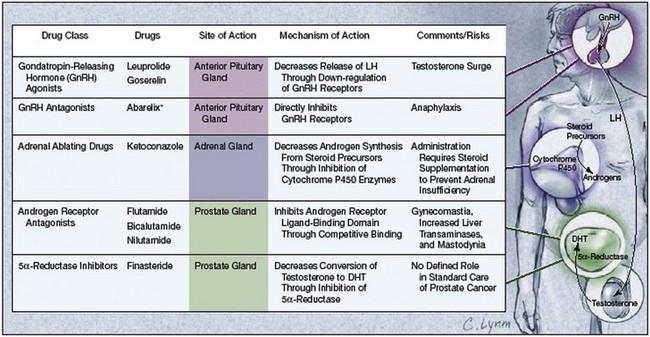
(From Sharifi N, Gulley JL, Dahut WL. Androgen deprivation therapy for prostate cancer. JAMA 2005;294:238–44.)
There are a variety of molecular mechanisms implicated in this process of androgen independence (Fig. 109–2). First, the AR pathway can become hypersensitive through a variety of molecular alterations and be activated by even lower levels of androgen (Linja et al, 2004). In androgen-refractory tumors, approximately one third of patients will show evidence of AR gene amplification, meaning many more copies of the AR gene are present (Koivisto et al, 1997; Linja et al, 2001). Second, the promiscuity of the AR for ligands other than androgen has been widely recognized. Mutations of the AR gene can also increase receptor activity (Tilley et al, 1996; Gottlieb et al, 1998; Taplin et al, 1999; Marcelli et al, 2000; Balk, 2002). Third, in an outlaw AR model, growth factor peptides, such as epithelial growth factor and insulin-like growth factor type 1 increase AR transcriptional activity in the absence of androgen (Culig et al, 1994). The cytokine interleukin-6 can activate AR, as can protein kinases A and C (Nazareth and Weigel, 1996; Lin et al, 2001). If these ligands can reactivate AR signaling in the absence of androgen, then prostate cancer can still progress in the castrate state. Increased expression of AR coregulators, proteins involved in the complex that binds to DNA, has been found in hormone-refractory prostate cancer, suggesting autonomous activation of the pathway (Yeh et al, 1999; Fujimoto et al, 2001; Gregory et al, 2001). Fourth, in a bypass AR model, activation of parallel or alternative survival pathways allows otherwise androgen-dependent prostate cancer cells to survive in the absence of androgen (Feldman and Feldman, 2001). Finally, in a lurker cell model, a small population of possible epithelial stem cells is preexistent in the prostate and androgen deprivation selects for the outgrowth of these androgen-independent cells. Rather than an acquired alteration, this model suggests an inherent quality of the prostate gland.
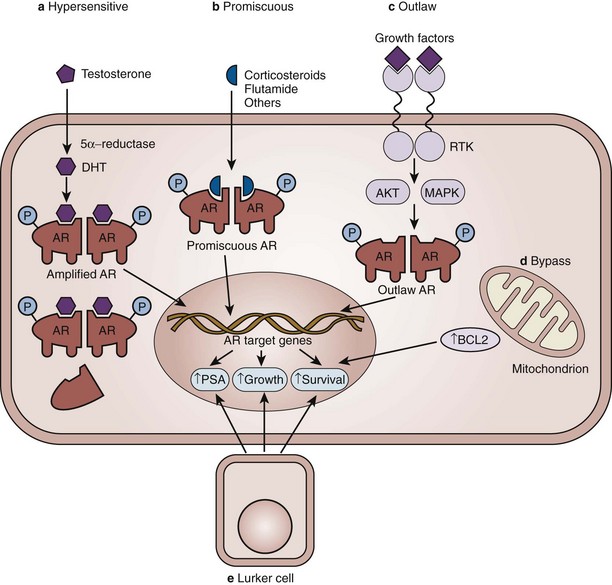
(From Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer 2001;1:34–45.)
Even when prostate cancer progresses despite castrate levels of androgen it is rarely resistant to androgen action. In 87% of patients with androgen-refractory prostate cancer, the administration of exogenous androgen results in symptomatic tumor flare (Fowler and Whitmore, 1982). Therefore the term androgen independent is not completely precise: such a cancer is no longer dependent on androgen (and thus may be considered independent), but since it remains responsive to androgen it is not wholly independent of the influence of androgen. The term androgen refractory is more precise, indicating a disease state in which the cancer is able to progress in the absence of androgen but is neutral about a responsive to androgen. Likewise, the word “hormone” is quite broad and includes the host factors defining the field of endocrinology. Therefore the term hormone independent is vague, as evident from the therapeutic response to secondary hormonal manipulation, estrogens, and glucocorticoids; and androgen-refractory prostate cancer is not actually independent of hormone action. The terms hormone independent and hormone refractory should be reserved for the rare cancers that are completely nonresponsive to any hormonal agent (Chang et al, 2005). Although hormone refractory prostate cancer is widely used to describe a state of progressive disease despite androgen-deprivation therapy, the term castration-resistant prostate cancer is more clinically precise and relevant (Scher et al, 2004, 2008).
Sources of Androgen
Testosterone is the major circulating androgen, with 90% produced by the testes. Over half of testosterone is bound to sex-binding globulin and 40% is bound to albumin. Only 3% of testosterone remains unbound, and this is the functionally active form of the hormone. After passive diffusion through the cell membrane into the cytoplasm, testosterone undergoes conversion to dihydrotestosterone (DHT) through the action of the enzyme 5α-reductase. Although the relative potency of testosterone and DHT are similar (as defined by the ability to cause half-maximal response in a prostate regrowth model), if the conversion of testosterone to DHT is blocked by the 5α-reductase inhibitor finasteride, 13-fold more testosterone is required for the same effect (Wright et al, 1999). Both testosterone and DHT exert their biologic effects by binding to the AR in the cytoplasm, promoting the association of AR coregulators. The complex then translocates to the nucleus and binds to androgen response elements in the promoter regions of target genes (Heinlein and Chang, 2004).
Androgens produced by the adrenal gland—androstenedione and dehydroepiandrosterone—are stimulated by adrenocorticotropic hormone (ACTH) released by the pituitary gland in response to corticotropic-releasing factor (CRF). Adrenal androgens negatively feed back on ACTH secretion: cortisol acts as the feedback signal. Adrenal androgens are relatively weak compared with testosterone and DHT and are almost entirely bound to albumin (Table 109–1). Adrenal androgens remain normal in men who have undergone orchiectomy (Walsh and Siiteri, 1975), and adrenal androgens are insufficient to maintain prostatic epithelium in such men.

Mechanisms of Androgen Axis Blockade
There are four therapeutic approaches for androgen axis blockade in current clinical use: (1) ablation of androgen sources, (2) inhibiting androgen synthesis, (3) antiandrogens, and (4) inhibition of LH-RH and/or LH release (Table 109–2).
Table 109–2 Therapeutic Approaches to Androgen-Deprivation Therapy: Several Agents Have Multiple Sites of Action

Ablation of Androgen Sources
Bilateral orchiectomy quickly reduces circulating testosterone levels to less than 50 ng/dL, which, based on this procedure, is considered the castrate range. Within 24 hours of surgical castration, testosterone levels are reduced by greater than 90% (Maatman et al, 1985). The Veterans Administration Cooperative Urological Research Group (VACURG) conducted a series of larger clinical trials, demonstrating the clinical effectiveness of surgical castration in reducing pain and performance status in men with advanced disease (Veterans Adminstration, 1967; Byar, 1973; Byar and Corle, 1988).
Scrotal (Simple) Orchiectomy
Subcapsular orchiectomy has been advocated as a technique of ADT that avoids the psychologic consequences of an empty scrotum (Desmond et al, 1988). Because this approach relies on the complete removal of all intratesticular tissue and Leydig cells it is more dependent on technique than a simple orchiectomy to achieve ADT. In a properly performed operation, however, the hormonal and cancer responses are indistinguishable from a simple, complete orchiectomy (Zhang et al, 1996).
Antiandrogens
Cyproterone Acetate
The classic steroidal antiandrogen with direct AR blocking effects, cyproterone acetate, also rapidly lowers testosterone levels to 70% to 80% through its progestational central inhibition (Jacobi et al, 1980; Goldenberg and Bruchovsky, 1991; Barradell and Faulds, 1994). An oral agent, the recommended dose is 100 mg, two to three times per day. Side effects are consistent with the hypogonadal state and include loss of libido, erectile dysfunction, and lassitude. Severe cardiovascular complications can occur in up to 10% of patients, limiting the use of this agent (deVoogt et al, 1986). Gynecomastia occurs in less than 20% of men. Rare cases of fulminate hepatotoxicity have been reported (Parys et al, 1991). Cyproterone acetate has been used at doses of 50 to 100 mg/day for the treatment of hot flashes (Goldenberg and Bruchovsky, 1991).
Nonsteroidal Antiandrogens
By blocking the testosterone feedback centrally the nonsteroidal antiandrogens cause LH and testosterone levels to increase. Testosterone levels reach about 1.5 times the normal levels of hormonally intact men (Neri, 1977). This allows antiandrogen activity without inducing hypogonadism: potency therefore can be preserved (Brufsky et al, 1997). However, in clinical trials specifically examining erectile functioning and sexual activity in men on flutamide monotherapy, long-term preservation of those domains was only 20%, not much different than men undergoing surgical castration (Schröder et al, 2000). The peripheral aromatization of increased testosterone to estradiol has been demonstrated after antiandrogen administration (Knuth, 1984), leading to the widely recognized gynecomastia and mastodynia associated with these agents. Gastrointestinal toxicity, and most notably diarrhea, is more common with flutamide than the other nonsteroidal antiandrogens (Han and Nelson, 2000). Liver toxicity, ranging from reversible hepatitis to fulminate hepatic failure, is associated with all nonsteroidal antiandrogens and requires periodic monitoring of liver function tests (Lund and Rasmussen, 1988; Wysowski et al, 1993; Dawson et al, 1997; Thole et al, 2004).
Antiandrogen Withdrawal Phenomenon
Patients treated with a combination of an antiandrogen and an LH-RH agonist can experience a decline in the prostate-specific antigen (PSA) value and even objective responses with the withdrawal of the antiandrogen from the combination. Based on this response, it appears the antiandrogen is actually exerting agonistic activity on prostate cancer cells. This phenomenon, first described with flutamide (Kelly and Scher, 1993), has now been demonstrated with all antiandrogens, including cyproterone acetate, as well as with DES and progestational agents (Kelly et al, 1997). Declines in PSA are seen within 4 weeks with flutamide withdrawal and within 6 weeks with bicalutamide and nilutamide withdrawal (Nieh, 1995). Between 15% and 30% of patients may have PSA level declines of greater than 50% after antiandrogen withdrawal with a median duration of 3.5 to 5 months (Scher and Kelly, 1993; Small and Srinivas, 1995). Objective, measurable tumor responses are observed less commonly. Overall survival has not been shown to be increased in those demonstrating the antiandrogen withdrawal phenomenon compared with those who have not (Small and Srinivas, 1995). Clinical trial designs of novel agents must take this phenomenon into consideration, given the possible confounding effects (Scher and Kelly, 1993). Prospective criteria to predict who will demonstrate this response have not been established, but it has been recognized that those with rapid PSA responses after androgen ablation have higher rates of antiandrogen withdrawal phenomenon.
It has been postulated that mutations in the AR may underlie this phenomenon, allowing the antiandrogen to behave as an activator of the AR (Taplin et al, 1995). The widely used prostate cancer cell line LNCaP expresses an AR with a specific point mutation that causes cell proliferation in the presence of hydroxyflutamide; the identical mutation was found in human tumor samples from patients who had remarkable declines in PSA levels after antiandrogen withdrawal (Suzuki et al, 1996). Similar point mutations in the AR have been described for bicalutamide to act as an agonist (Hara et al, 2003), with the structural basis of this mutation resolved by x-ray crystallography demonstrating the ability of bicalutamide to bind to the mutant AR in a fashion similar to DHT to the wild-type AR (Bohl et al, 2005).
Flutamide
A nonsteroidal antiandrogen, flutamide was the first “pure” antiandrogen (Neri et al, 1967). Because of the short half-life (6 hours) of the active metabolite, 2-hydroxyflutamide, this oral agent is given in a dose of 250 mg three times a day. Elimination of hydroxyflutamide is via renal excretion. Unlike the steroidal antiandrogens there are no associated side effects of fluid retention or thromboembolism (Dalaere and Van Thillo, 1991). In a randomized, double-blind study comparing flutamide to DES (3 mg/day) in metastatic prostate cancer, overall survival was significantly shorter with flutamide (28.5 months) compared with DES (43.2 months) (Chang et al, 1996).
Bicalutamide
A nonsteroidal antiandrogen with a long serum half-life (6 days), bicalutamide has a once-per-day dosing schedule and therefore likely better compliance. It is the most potent of the nonsteroidal antiandrogens (Kolvenbag and Nash, 1999) and the best tolerated (Kolvenbag and Blackledge, 1996; Schellhammer and Davis, 2004; Fradet, 2004). The pharmacokinetics of bicalutamide are not affected by age, renal insufficiency, or moderate hepatic impairment (Mahler et al, 1998). The R-isomer of bicalutamide has about a 30-fold higher binding affinity to the AR compared with the S-isomer and functionally processes the antiandrogen activity (Mukherjee et al, 1996). As with the other antiandrogens, bicalutamide is associated with maintenance of serum testosterone levels: in the majority of patients these remain within the normal range (Denis and Mahler, 1996; Tay et al, 2004).
Bicalutamide as monotherapy has been most extensively studied and, as with flutamide monotherapy’s inferiority to DES, bicalutamide monotherapy at a 50-mg/day dose was inferior to castration in survival of men with metastatic disease (Kaisary et al, 1995; Bales and Chodak, 1996; Kolvenbag and Nash, 1999). At higher dose of 150 mg/day, however, bicalutamide monotherapy appears to have equivalent efficacy to medical or surgical castration (Tyrrell et al, 1998; Iversen et al, 2000; Anderson, 2003; Iversen et al, 2004; Wirth et al, 2004, 2005; McLeod et al, 2005) in men with metastatic or locally advanced disease. In these large phase 3 studies, bicalutamide monotherapy (150 mg/day) had significantly better quality of life in the domains of sexual interest and physical capacity (Iversen, 2003). There was, however, a high rate of gynecomastia (66.2%) and breast pain (72.8%) (Iversen, 2003). Of more concern, in men with low-risk, localized prostate cancer, bicalutamide was associated with significantly worse overall survival compared with those on “watchful waiting” (see later).
Nilutamide
The plasma half-life of nilutamide is 56 hours, and elimination is via hepatic clearance employing the cytochrome P450 system. Because steady-state plasma levels are achieved in 14 days on once-daily dosing (Creaven et al, 1991), dosing recommendations are a single 300-mg daily dose for the first month of treatment followed by a single 150-mg daily dose (Mahler et al, 1998). About one fourth of men on nilutamide therapy will note a delayed adaptation to darkness after exposure to bright illumination (Creaven et al, 1991). Nilutamide is also associated with interstitial pneumonitis in approximately 1% of patients, which can progress to pulmonary fibrosis (Pfitzenmeyer et al, 1992). The early effects are usually reversible with cessation of nilutamide. In a small study there was a suggestion of a role for nilutamide as an effective secondary hormonal agent (Desai et al, 2001).
MDV3100
This small molecule is an AR antagonist, but, unlike bicalutamide, MDV3100 inhibits AR function by blocking nuclear translocation and DNA binding and has no agonist activity when AR is overexpressed. It is being evaluated in phase 1/2 clinical trials in men with castration-resistant prostate cancer. Early results indicate the PSA level declines in the majority of patients (Scher et al, 2008).
Inhibition of LH-RH
LH-RH Agonists
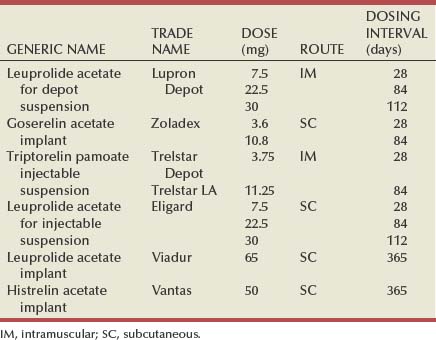
The LH-RH agonists exploit the desensitization of LH-RH receptors in the anterior pituitary after chronic exposure to LH-RH, thereby shutting down the production of LH and, ultimately, testosterone. The clinical utility of the current LH-RH agonists is based on the creation of analogues of native LH-RH by amino acid substitutions, particularly position 6 in the peptide, increasing their potency and half-lives (Table 109–3). Pharmacologic depot preparations and osmotic pump devices allow dosing to extend from 28 days to 1 year, respectively (Table 109–4) (Ahmann et al, 1987). In a review of 24 trials, involving more than 6600 patients, survival after therapy with an LH-RH agonist was equivalent to that of orchiectomy (Seidenfeld et al, 2000).
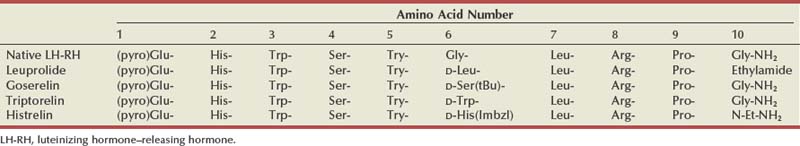
Table 109–4 Luteinizing Hormone–Releasing Hormone Agonists Approved for the Treatment of Prostate Cancer
The initial exposure to more potent agonists of LH-RH results in a flare of LH and testosterone levels (Waxman et al, 1985). This phenomenon is seen with all available LH-RH preparations and can result in a severe, life-threatening exacerbation of symptoms. The flare, associated with up to a 10-fold increase in LH, may last for 10 to 20 days (Weckerman and Harzmann, 2004). Fortunately, the coadministration of an antiandrogen functionally blocks the increased levels of testosterone (Labrie et al, 1987; Kuhn et al, 1989; Schultz and Senge, 1990). Although it had been argued that the administration of the antiandrogen should precede the administration of the LH-RH agonist by a week, others have found no differences in PSA levels with the simultaneous administration of both agents (Tsushima et al, 2001). Given the predictable length of the flare phenomenon, antiandrogen coadministration is required for only 21 to 28 days.
LH-RH Antagonists
The LH-RH antagonists bind immediately and competitively to the LH-RH receptors in the pituitary, reducing LH concentrations by 84% within 24 hours of administration (Weckerman et al, 2004). The direct antagonistic activity eliminates the LH and testosterone flare, which is the major therapeutic advantage of these agents: there is no need for antiandrogen coadministration. Hormonally naive patients with impending spinal cord compression or severe bone pain for whom surgical castration is not appropriate may uniquely benefit from this class of agents: clinical response has been observed with the LH-RH antagonist cetrorelix (Gonzalez-Barcena et al, 1995).
In clinical trials of the LH-RH antagonist abarelix, testosterone levels dropped very quickly, with 34.5%, 60.5%, and 98.1% of men chemically castrated at 2, 4, and 28 days, respectively (Tomera et al, 2001). Compared with an LH-RH agonist and an antiandrogen, abarelix monotherapy was equally effective in achieving castrate levels of testosterone (Trachtenberg et al, 2002). Ninety-percent of men with symptomatic prostate cancer treated in an open-label fashion had improvements in pain and/or disease-related problems (Koch et al, 2003).
Many of the first- and second-generation antagonists induced significant histamine-mediated side effects, complications not as often observed in third- and fourth-generation agents (Weckerman et al, 2004). Nevertheless, severe allergic reactions can occur with abarelix, even after previously uneventful treatment (Koch et al, 2003). Abarelix is approved in the United States for the treatment of advanced prostate cancer in patients who cannot take other hormonal therapies and have refused surgical castration. Given the rare but serious allergic reactions, patients must be monitored for at least 30 minutes after administration.
FSH levels are only partially suppressed by LH-RH agonists and are significantly elevated after surgical castration given the loss of inhibitory feedback. LH-RH antagonists reduce both LH and FSH levels. In an androgen-insensitive prostate cancer xenograft model, cetrorelix significantly reduced tumor growth (Lamharzi et al, 1998), suggesting other factors stimulate tumor growth. In men with disease progression after surgical castration, treatment with abarelix reduced FSH levels by nearly 90% but did not meet criteria for PSA response (Beer et al, 2004a).
A new LH-RH antagonist, degarelix, has completed phase 2 and phase 3 studies in prostate cancer (Gittelman et al, 2008; Klotz et al, 2008). Unlike with abarelix there were no systemic allergic reactions with degarelix. In the phase 3 study, degarelix was compared with leuprolide: at 1 year of treatment degarelix was not inferior to leuprolide, and, based on this result, it has been approved for use in the United States.
Inhibition of Androgen Synthesis
Aminoglutethimide
Aminoglutethimide inhibits the conversion of cholesterol to pregnenolone, an early step in steroidogenesis (Cash et al, 1967; Blankenstein and Bakker, 1985). Given its inhibition of a very proximal step in adrenal function, aminoglutethimide blocks production of aldosterone and cortisol. As the medical version of a total adrenalectomy, the use of this agent requires replacement of cortisone and fludrocortisone. Side effects include anorexia, nausea, rash, lethargy, vertigo, hypothyroidism, and nystagmus. Clinical responses have been observed in a subset of patients with androgen-refractory prostate cancer treated with aminoglutethimide plus cortisone (Sanford et al, 1976; Ponder et al, 1984). In the PSA era, 37% of patients had greater than a 50% decline in the PSA level with treatment with aminoglutethimide (1000 mg/day) and hydrocortisone acetate (40 mg/day), with median response times lasting 9 months (Kruit et al, 2004).
Ketoconazole
An orally active, broad-spectrum azole antifungal agent, ketoconazole interferes with two cytochrome P450–dependent pathways: by inhibiting 14-methylation the conversion lanosterol to cholesterol is blocked, and it also blocks 17,20-desmolase, impacting on the conversion of C21 to C19 steroids. Based on the observation that some patients taking the drug developed gynecomastia, investigations on its effects on steroid synthesis demonstrated loss of adrenal steroid synthesis (Pont et al, 1982b) and testosterone synthesis by Leydig cells (Pont et al, 1982a). The effects were rapid, with testosterone levels dropping to the castrate level within 4 hours of administration in some cases (Trachtenberg et al, 1983); the effects were also immediately reversible, indicating dosing must be continuous to maintain low testosterone levels (400 mg every 8 hours).
Early experience with ketoconazole in the treatment of prostate cancer showed this agent to be tolerable, durable, and effective (Trachtenberg and Pont, 1984) and was palliative for those who had failed first-line androgen-ablation therapy (Pont, 1987). Although effective in rapidly bringing testosterone levels into the castrate range, with continuous treatment with ketoconazole in the otherwise hormonally intact individual (no other surgical or chemical ADT), testosterone levels begin to rise and can reach low-normal ranges within 5 months of therapy (Vanuytsel et al, 1987). Therefore currently, ketoconazole is used for men with castration-resistant prostate cancer, often as the first or second agent in so-called secondary hormonal manipulation (Small et al, 2004). In addition to gynecomastia (caused by alterations in testosterone-estradiol ratios [Pont et al, 1985]), ketoconazole is associated with lethargy, weakness, hepatic dysfunction, visual disturbance, and nausea (Wilkinson and Chodak, 2004; Scholz et al, 2005). Because of the adrenal suppression, ketoconazole is usually given with hydrocortisone (20 mg twice daily). Patients with higher levels of the adrenal androgen androstenedione had a improved survival in response to ketoconazole compared with those with lower levels, suggesting ketoconazole is less effective in patients with low levels of androgen at baseline (Ryan et al, 2007).
Abiraterone
Unlike the less potent ketoconazole, which inhibits several cytochrome P pathways, abiraterone is a potent, selective and irreversible inhibitor of cytochrome P17, a key enzyme in androgen synthesis (Chan et al, 1996). Specifically, abiraterone inhibits 17α-hydrolase, resulting in excess synthesis of aldosterone and its precursors and causing a suppression of cortisol with a compensatory rise in ACTH. Abiraterone also inhibits C17,20-lyase, resulting in suppression of testosterone to levels of less than 1 ng/mL (significantly < 50 ng/ml, castrate levels) (O’Donnell et al, 2004; Attard et al, 2005). Based on the hypothesis that castration-resistant prostate cancer remains driven by low levels of androgens, abiraterone is undergoing clinical testing in this state of disease. In a phase 1 clinical trial, abiraterone was well tolerated, with toxicities associated with the expected effects of mineralocorticoid excess, namely, hypertension, hypokalemia, and lower-extremity edema (Attard et al, 2008). Declines in PSA level and measurable tumor response were observed and supported by preliminary phase 2 results (de Bono et al, 2008). Phase 3 studies are underway.
Response to Androgen Blockade
After the initiation of ADT the vast majority of prostate cancer patients will show some evidence of clinical response: the magnitude and rapidity of that response remain the best predictors of its durability. Assuming ADT effectively targets the androgen-sensitive population of prostate cancer cells, an incomplete or sluggish response is evidence of a significant androgen-refractory population. Early in the clinical use of PSA as a biomarker of prostate cancer it was recognized that declines in PSA could predict response (Hudson et al, 1989; Aria et al, 1990; Cooper et al; 1990). For example, patients who had more than an 80% drop of PSA within 1 month of initiating ADT had a significantly longer disease-free progression rate (Aria et al; 1990). Likewise, the nadir PSA predicted the progression-free interval (Matzkin et al, 1992; Benaim et al, 2002a), as did pretreatment testosterone levels (Imamoto et al, 2001). A rise in PSA level, which is evidence of the emergence of castration-resistant disease, preceded bone metastatic progression by several months, with a mean lead time of 7.3 months (Cooper et al, 1990; Miller et al, 1992).
More recent studies of PSA response to ADT have confirmed and amplified those observations. The odds ratio for progressing to castration-resistant progression with 24 months of starting ADT was almost 15 times higher for patients who did not achieve an undetectable PSA level (Benaim et al, 2002a). For each unit increase in Gleason score, the cumulative hazard of castration-resistant progression was nearly 70% (Benaim et al, 2002b). In one cohort of Asian men the nadir PSA was the most accurate predictor of disease progression and was independently prognostic of survival; achieving a PSA level of 1.1 ng/mL or less at 6 months after initiating ADT was the most sensitive and specific predictor of progression at 2 years (Kwak et al, 2002). In men with newly diagnosed metastatic prostate cancer (stage D2) started on ADT the absolute PSA level after 7 months of ADT was a strong, independent predictor of survival (Hussain et al, 2006). Median survival of men achieving a PSA level less than or equal to 0.2 ng/mL was 75 months, compared with 13 months for men whose nadir was more than 4 ng/mL (Fig. 109–3). Considering the kinetics of PSA rise before ADT compared with the rate of PSA decline after ADT also predicted outcome, specifically prostate cancer–specific mortality (D’Amico et al, 2005). If the pre-ADT PSA rise was rapid and the decline after ADT was slow the cancer-specific mortality was significantly worse compared with those with slow rises in PSA before ADT and rapid declines after ADT (D’Amico et al, 2004a, 2004b).
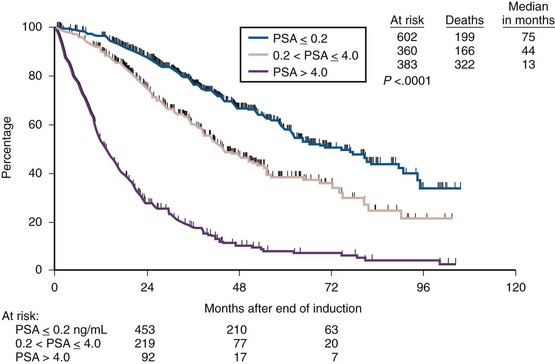
(From Hussain M, Tangen CM, Higano C, et al. Absolute prostate-specific antigen value after androgen deprivation is a strong independent predictor of survival in new metastatic prostate cancer: data from Southwest Oncology Group Trial 9346 (INT-0162). J Clin Oncol 2006;24:3984–90.)
Almost without exception those no longer responding to ADT (androgen-refractory) remain on ADT. Therefore factors influencing survival in that disease state should be considered in this discussion. In most cases, available data are based on pre- or post-treatment responses to other systemic treatments (Galsky and Kelly, 2003). Consistently predictive variables (by both univariate and multivariate analysis) of survival in this state include performance status; serum lactate dehydrogenase (LDH), serum alkaline phosphatase, and hemoglobin levels; and PSA response to secondary therapy (Smalez et al, 2002). The survival of men treated on seven sequential chemotherapy protocols at one institution provided an early experience in developing predictive measures (Kelly et al, 1993). A 50% decline in PSA value in response to chemotherapy was one of the most significant variables predicting survival. A nomogram, based on a larger groups of patients, finds the presence of visceral disease, Gleason score, performance status, baseline PSA value, LDH and alkaline phosphatase levels, and hemoglobin level were useful in modeling prognosis (Smalez et al, 2002; Halabi et al, 2003).
General Complications of Androgen Ablation
Osteoporosis
The increased number of men being placed on androgen ablation therapy much earlier in the course of their disease allows the chronic manifestations of the hypogonadal state to emerge. Widespread androgen ablation therapy applied to an increasing aging population, already predisposed to loss of bone mineral density (BMD), has created an epidemic of osteopenia and osteoporosis. Fragile bones increase the risk of skeletal fracture. More than half of men meet the BMD criteria for osteopenia or osteoporosis—defined as more than 2.5 standard deviations below an age-specific reference mean—before the initiation of ADT (Wei et al, 1999; Conde et al, 2004). The longer a man remains on ADT, the greater the risk of fracture (Daniell et al, 2000; Krupski et al, 2004). After 5 years on ADT, 19.4% of men experienced fractures compared with 12.6% of controls (Shahinian et al, 2005): over 15 years, cumulative incidence of fractures was 40%, compared with 19% in noncastrate controls (Melton et al, 2003). It has been estimated that 4 years of ADT will place the average man in the osteopenic range (Wei et al, 1999). Prostate cancer was associated with an increased odds ratio (95% confidence intervals) for hip fractures of 3.7 (3.1 to 4.4) in a large Danish population-based study (Abrahamsen et al, 2007). Rarely discussed even 10 years ago, skeletal health is now becoming a major concern of patients and their physicians (Chen et al, 2002).
Treatment of osteoporosis begins with recognition. BMD of the hip, as measured by dual-energy x-ray absorptiometry, should be considered on all men who are anticipated to be on long-term ADT (Bae and Stein, 2004; Diamond et al, 2004). Smoking cessation, weight-bearing exercise, and vitamin D and calcium supplementation can help improve BMD. Daily supplementation of calcium and vitamin D is recommended by the National Institutes of Healt h at doses of 1200 to 1500 mg/day and 400 IU/day, respectively (Michaelson et al, 2008). Supplementation decreases the incidence of nonvertebral fractures in men and women older than 65 years of age (Dawson-Hughes et al, 1997). Prevention of osteoporosis in men receiving ADT has been demonstrated in controlled studies using the bisphosphonate pamidronate (Smith et al, 2001). In a randomized prospective study, once-weekly oral alendronate reversed bone loss associated with ADT and improved bone density; this benefit continued with ongoing treatment, suggesting chronic treatment is beneficial (Greenspan et al, 2007, 2008). BMD also increased in men on ADT using the considerably more potent bisphosphonate zoledronic acid (Smith et al, 2003a). Bisphosphonate therapy should be considered in any man with evidence of osteopenia or osteoporosis (Bae and Stein, 2004). Transdermal estradiol also increases BMD in men with prostate cancer (Ockrim et al, 2004). Not surprisingly, serum testosterone and estradiol levels were much lower on men receiving LH-RH agonists compared with those on nonsteroidal antiandrogen therapy; interestingly, markers of bone turnover were significantly higher in men taking LH-RH agonists compared with those on nonsteroidal antiandrogen therapy, suggesting the nonsteroidal antiandrogens may help maintain BMD (Smith et al, 2003b).
Hot Flashes
For more than 100 years, hot flashes (also called hot flushes, vasomotor symptoms) have been recognized as a side effect of androgen ablation: in 1896 Cabot mentioned “uncomfortable flushes of heat, similar to those experienced by women at the time of menopause” in men undergoing castration for prostatic enlargement (Cabot, 1896; Stearns, 2004). Described as a subjective feeling of warmth in the upper torso and head followed by objective perspiration, hot flashes are not life threatening but are among the most common side effects of androgen ablation, affecting between 50% and 80% of patients (Moyad, 2002; Spetz et al, 2003; Nishiyama et al, 2004). Occurring both spontaneously and precipitated by changes in body position, ingestion of hot liquids, or changes in environmental temperature, the exact etiology of hot flashes remains undefined. Among the proposed mechanisms include increases in hypothalamic adrenergic concentrations and alterations in β-endorphins and calcitonin gene–related peptides acting on the thermoregulatory center in the hypothalamus (Yuzurihara et al, 2003). Hot flashes generally decrease both in frequency and intensity over time but often persist in some men (Holzbeierlein et al, 2004).
Treatment of hot flashes should be reserved for those who find them bothersome. Just as hot flashes are a consequence of alterations in the hormonal milieu the mainstay of treatment has been based on efforts to influence that milieu (Kouriefs et al, 2001). In a double-blind, placebo-controlled, crossover study, the progestational agent megestrol acetate (20 mg, twice per day) significantly reduced the frequency of hot flashes (Loprinzi et al, 1994). The dose can be reduced to 5 mg twice daily, which may help reduce the appetite-stimulating effect of this agent. The efficacy of cyproterone acetate is based on its progestational effects (Cervenakov et al, 2000). Dosing should start at 50 mg/day and be titrated to 300 mg/day. Estrogenic compounds, such as low-dose DES and transdermal estradiol, appear to be the most effective treatment, with up to 90% partial or complete resolution of symptoms (0; Smith, 1994; Gerber et al, 2000). With estrogen compounds, however, the cure may be worse than the disease; painful gynecomastia and thromboembolic effects have limited the utility of this approach. Clonidine, a centrally acting α agonist that decreases vascular reactivity, has been used with mixed results: in a placebo-controlled study, transdermal clonidine did not significant decrease hot flashes (Loprinzi et al, 1994). Antidepressant agents, particularly the selective serotonin reuptake inhibitor venlafaxine (12.5 mg, twice daily), have reduced hot flashes in more than 50% of men (Quella et al, 1999; Loprinzi et al, 2004). In a phase 3 randomized, double-blind, placebo-controlled trial, the antiseizure agent gabapentin decreased hot flashes to a moderate degree (Loprinzi et al, 2009).
Sexual Dysfunction (Erectile Dysfunction and Loss of Libido)
The effects of ADT on sexual function are profound, as first described by Huggins and associates (1941): “Sexual desire and penile erections were absent in all cases following castration.” Loss of sexual functioning is not inevitable, however, with up to 20% of men on ADT able to maintain some sexual activity (Rousseau et al, 1988; Clark et al, 2001). Specifically, between 10% and 17% of men undergoing ADT can maintain an erection adequate for intercourse (Tomic, 1983; Potosky et al, 2001). Libido is more severely compromised, with approximately 5% of men maintaining a high level of sexual interest with ADT (Potosky et al, 2001). Sexual desire is inversely related to the duration of androgen deprivation (Basaria et al, 2002). Loss of penile volume, penile length, nocturnal penile tumescence, and, for those undergoing medical ADT, testicular volume is common (Marumo et al, 1999; Higano, 2003).
Treatment for loss of libido is extremely difficult—if not impossible—for those on ADT. Likewise, medical treatments, such as oral phosphodiesterase type 5 inhibitors, or local treatments, such as intracavernosal injections of alprostadil, can still be effective in selected patients but may not be used over the long term. If there is any fairness in the negative effects of ADT on sexual function it is the decline in both libido and erectile functioning: despite no erections or desire, the majority of patients have little to no problem with their lack of sexual functioning (Potosky et al, 2001).
Cognitive Function
There is a strong suggestion that ADT is linked to subtle but significant cognitive declines in men with prostate cancer (Nelson et al, 2008). In both men and women the hypogonadal state is associated with declines in cognitive functioning (Gouchie and Kimura, 1991; Sherwin and Tulandi, 1996). Testosterone supplementation improves verbal fluency (Alexander et al, 1998) whereas other controlled studies have found no effect of such supplementation on memory (Sih et al, 1997). In one small study, men with prostate cancer randomized to ADT performed worse in cognitive studies compared with men with prostate cancer under surveillance (Green et al, 2002); the declines were associated with tasks requiring complex information processing (Green et al, 2004). Compared with tests for other cognitive domains, tests for spatial ability uniquely declined in men on intermittent hormone therapy (Cherrier et al, 2003). In men on neoadjuvant ADT before radiotherapy, cognitive functioning declined (Jenkins et al, 2005). Unfortunately, the studies examining the effects of ADT on cognitive functioning have been small and underpowered. Not surprisingly, given many side effects of ADT, quality of life worsens, specifically in men receiving flutamide in addition to castration, compared with placebo, in the domain of emotional functioning (Moinpour et al, 1998). A short course of ADT (36 weeks) increased depression and anxiety scores on formal neuropsychologic evaluations (Almeida et al, 2004); major depressive disorder was prevalent in 12.8% of men on ADT, 8 times greater than the national rate and 32 times the rate of men older than 65 years of age (Pirl et al, 2002). Finally, psychologic distress accounted for approximately one third of declines in fatigue severity scale in men undergoing ADT (Stone et al, 2000).
Changes in Body Habitus
A loss of muscle mass and increase in percent fat body mass are common in men undergoing ADT, and these changes are most pronounced with the initiation of ADT (van Londen et al, 2008
Related posts:
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Prosthetic Surgery for Erectile Dysfunction
Prosthetic Surgery for Erectile Dysfunction
 Urodynamic and Video-Urodynamic Evaluation of the Lower Urinary Tract
Urodynamic and Video-Urodynamic Evaluation of the Lower Urinary Tract
 Neuropathic Dysfunction of the Lower Urinary Tract
Neuropathic Dysfunction of the Lower Urinary Tract
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


