Essentials of Diagnosis
General Considerations
Renal dysfunction is a common and serious problem in patients with advanced liver disease, estimated to occur in 10% of hospitalized patients with cirrhosis. It is a syndrome characterized by (1) oliguria, severe renal sodium retention, and rapidly progressive azotemia, (2) circulatory instability with marked systemic arterial vasodilation and activation of vasoactive systems, and (3) a very poor prognosis. Without treatment, the median survival for type 1 hepatorenal syndrome patients is on the order of 1–2 weeks, while that for type 2 hepatorenal syndrome is about 20% at 1 year. However, hepatorenal syndrome has always been considered to be a form of functional renal failure, as kidneys from patients with hepatorenal syndrome, when transplanted into someone with renal failure, regain their normal function. Similarly, renal function also improves in patients with end-stage cirrhosis following liver transplantation, although the renal function can remain abnormal for quite sometime in the postoperative period.
Hepatorenal syndrome is defined as the development of renal failure in patients with advanced liver failure (acute or chronic) in the absence of any identifiable causes of renal pathology. It is a diagnosis of exclusion, when all other causes of renal failure, functional or organic, have been excluded. The International Ascites Club further defines the criteria for the diagnosis of hepatorenal syndrome, as detailed in Table 10–1. It must be emphasized that urinary parameters are supportive, but not essential for the diagnosis of hepatorenal syndrome. For example, urinary volume is usually <500 mL/day, but there are nonoliguric forms of hepatorenal syndrome. Urinary sodium excretion is usually <10 mEq/day in hepatorenal syndrome. However, cases of well-documented hepatorenal syndrome with urinary sodium of >10 mEq/day have been reported. Finally, although the urinary osmolality is higher than the plasma osmolality in most patients with hepatorenal syndrome, a decrease in urinary osmolality may occur as renal failure progresses.
Cirrhosis with ascites |
Serum creatinine >133 μmol/L (1.5 mg/dL) |
No improvement of serum creatinine (decrease to a level of ≤133 μmol/L) after at least 2 days of diuretic withdrawal and volume expansion with albumin; the recommended dose of albumin is 1 g/kg body weight/day up to a maximum of 100 g/day |
Absence of shock |
No current or recent treatment with nephrotoxic drugs |
Absence of parenchymal kidney disease as indicated by proteinuria >500 mg/day, microhematuria (>50 red blood cells per high power field), and/or abnormal renal ultrasonography |
The Internal Ascites Club divided hepatorenal syndrome into type 1 and type 2. Type 1 hepatorenal syndrome is characterized by a rapid decline in renal function defined as a doubling of serum creatinine to a level greater than 220 μmol/L or a halving of the creatinine clearance to less than 20 mL/minute within 2 weeks. The clinical presentation is that of acute renal failure. The patient is usually very sick, with marked jaundice and severe coagulopathy. In type 2 hepatorenal syndrome, renal function deteriorates more slowly with the serum creatinine increasing to greater than 133 μmol/L or a creatinine clearance decreasing to less than 40 mL/minute over the course of weeks to months. The clinical presentation is that of gradual renal failure in a patient with cirrhosis and refractory ascites.
Pathogenesis
The pathophysiology of hepatorenal syndrome is complex. The hallmark of hepatorenal syndrome is renal hypoperfusion, which is due to a reduction in the renal perfusion pressure as well as to active renal vasoconstriction. This leads to a decrease in the renal blood flow and a reduction in the glomerular filtration rate. Many pathophysiologic factors are involved in the development of hepatorenal syndrome in patients with end-stage cirrhosis.
Liver cirrhosis and portal hypertension are characterized by an increased cardiac output and decreased systemic vascular resistance, the so-called hyperdynamic circulation. The basis for the hyperdynamic circulation is systemic arterial vasodilation. This occurs mainly in the splanchnic circulation, a result of both increased resistance to portal flow due to the cirrhosis and to the presence of excess vasodilators and/or decreased responsiveness of the vasculature to endogenous vasoconstrictors. Clinically, this is manifested as systemic hypotension, tachycardia, wide pulse pressure, and warm peripheries. The homeostatic response is the activation of various vasoconstrictor systems, including the renin–angiotensin system, the sympathetic nervous system, and arginine vasopressin. These will counteract the vasodilatory effects of the vasodilators and direct the kidneys to retain sodium and water in order to maintain hemodynamic stability. As cirrhosis progresses, systemic hypotension worsens as the systemic arterial vasodilation increases, and at some point in time the renal perfusion pressure will fall. When combined with increasing levels of the systemic vasoconstrictors, total renal blood flow gradually falls. When the production of endogenous vasodilators cannot keep pace with the fall in renal blood flow, renal failure ensues (Figure 10–1).
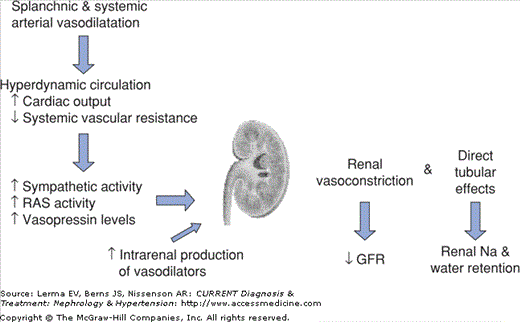
Not all cirrhotic patients with similarly reduced renal blood flow will develop hepatorenal syndrome. Therefore, other factors must also play a pathogenetic role. In addition to reduced renal blood flow, various vasoconstrictors, in particular, endothelin-1 and leukotrienes, may also cause mesangial contraction, thereby reducing the glomerular ultrafiltration coefficient and further decreasing the glomerular filtration rate.
Portal hypertension is associated with a reduction of renal blood flow and may play a role in the pathogenesis of hepatorenal syndrome. The connection between portal hypertension and renal hemodynamics may be the sympathetic nervous system.
Renal hypoperfusion, due to a reduction of renal vasodilators, in cirrhosis could also be related to liver dysfunction. However, the mechanism whereby liver dysfunction could directly induce a reduction in renal vasodilators is unclear. It is possible that the liver is involved in the synthesis or release of renal vasodilators such as nitric oxide. The severe jaundice associated with liver failure can sensitize the renal vasculature to the vasoconstrictive effects of norepinephrine, resulting in greater renal vasoconstriction with any given level of circulating norepinephrine. High levels of bile acids observed in cholestasis can cause arterial vasodilation, thereby exaggerating the hemodynamic instability. That is, hepatic dysfunction “makes a bad situation worse.” In patients with severe jaundice (bilirubin >510 μmol/L), there is direct bilirubin nephrotoxicity.
At least 50% of patients with hepatorenal syndrome arrive in the hospital with near normal renal function. Therefore, it is what clinicians do to these patients that frequently precipitates hepatorenal syndrome. These precipitating events cause a further reduction in the filling of the effective arterial circulation, exaggerating the hemodynamic instability, resulting in further renal hypoperfusion and a decrease in the glomerular filtration rate.
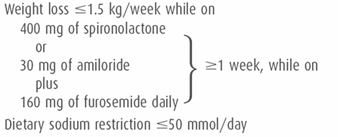
Cirrhotic patients with refractory ascites (Table 10–2) experience a reduction in their effective arterial blood volume and do not respond well to diuretic therapy. Diuretic therapy, by decreasing the intravascular volume, further exaggerates the reduction in effective arterial blood volume, and predisposes the patient to the development of hepatorenal syndrome. Clinicians have a tendency to increase the diuretic doses when there is an inadequate diuretic and natriuretic response, despite rising serum creatinine levels. Cirrhotic patients with refractory ascites typically excrete only approximately 500 mL of urine per day even in the presence of a “normal” serum creatinine level. Therefore, when increasing doses of diuretics do not result in an increased urine volume or urinary sodium excretion, further increases in the diuretic doses will increase the likelihood of developing hepatorenal syndrome in these patients. Conversely, decreasing the diuretic doses in a patient with refractory ascites and rising serum creatinine may reverse the renal dysfunction.
Large volume paracentesis leads to an exaggeration of the hyperdynamic circulation. The systemic circulation becomes more vasodilated about 24 hours after a large volume paracentesis. The subsequent further activation of the vasoconstrictor systems predisposes the patient to the development of hepatorenal syndrome. Reducing the rate of paracentesis could potentially prevent deleterious hemodynamic consequences and reduce the risk of developing hepatorenal syndrome.
It is estimated that at least 30% of patients with spontaneous bacterial peritonitis will develop hepatorenal syndrome despite adequate treatment of their infection. It has been postulated that sepsis in cirrhosis induces an increased production of various cytokines and endotoxins, which in turn stimulates the production of nitric oxide and other vasodilators, causing further arterial vasodilation. Therefore, spontaneous bacterial peritonitis exaggerates the reduction in the effective arterial blood volume and increases the risk of further deterioration of the systemic hemodynamics, leading to decreased renal function.
Acute blood loss with acute blood volume contraction usually leads to acute tubular necrosis and not hepatorenal syndrome. However, patients with decompensated cirrhosis and gastrointestinal bleeding can develop a systemic inflammatory response syndrome, manifesting as an increase in temperature, tachycardia, tachypnoea, and leukocytosis with or without an infection, associated with the activation of many cytokines. Once again, these cytokines can stimulate the production of nitric oxide and other vasodilators. Thus, the patient with gastrointestinal bleeding is also predisposed to further exaggeration of the systemic arterial vasodilation, due to the fact that the accompanying inflammatory response will yield more vasodilators, aggravating the effective arterial underfilling. Gastrointestinal bleeding also predisposes the cirrhotic patient to the development of infection, which in turn predicts rebleeding after control of the initial bleeding episode. The presence of infection in a cirrhotic patient with gastrointestinal bleeding adds to the inflammatory response and to cytokine production, further exaggerating the hemodynamic instability and increasing the likelihood of developing hepatorenal syndrome. To support this hypothesis, the routine use of prophylactic antibiotics in cirrhosis with gastrointestinal bleeding has led to a significant reduction in the incidence of hepatorenal syndrome associated with the bleeding episode.
Acute biliary obstruction is associated with the development of renal impairment, leading to the suggestion that increased production of F2-isoprostanes in cholestasis was responsible for the development of renal failure, since F2-isoprostanes are potent renal vasoconstrictors, and the administration of antioxidants that reduced F2-isoprostane levels was associated with improvement of renal function. Cholestasis per se appears to be detrimental to the systemic circulation. It is not surprising that when cholestasis is superimposed on cirrhosis and portal hypertension, the circulatory changes are likely to worsen, predisposing the patient to the development of hepatorenal syndrome.
The use of nonselective nonsteroidal anti-inflammatory drugs (NSAIDs) in cirrhosis is associated with a reduction in renal perfusion and glomerular filtration rate, secondary to an inhibition of the renal production of vasodilatory prostaglandins. In addition, NSAIDs impair renal sodium and water excretion, and these effects are independent of the deterioration in renal hemodynamics. These effects are particularly pronounced in cirrhosis with ascites, as these patients are dependent on the intrarenal production of prostaglandins to counteract the effects of various vasoconstrictors. Therefore, ascitic cirrhotic patients should not receive NASIDs in order to avoid the precipitation of hepatorenal syndrome. Cirrhotic patients are dependent on the activated renin–angiotensin system to maintain systemic blood pressure. Therefore, the use of angiotensin-converting enzyme inhibitors and angiotensin II antagonists can result in arterial hypotension and precipitate renal failure in these patients.
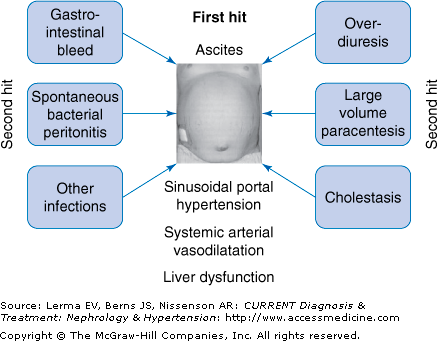
The pathogenesis of hepatorenal syndrome can be best summed up by the 2-hit theory (Figure 10–2). The cirrhotic patient with advanced liver disease and massive ascites has a compromised circulatory state (the first hit). The activation of various compensatory mechanisms maintains the circulation. Further progression of liver disease with deterioration of the circulation will lead to the development of hepatorenal syndrome (nonprecipitated cases). Alternatively, the presence of a precipitating factor will lead to rapid deterioration of the systemic circulation and to the development of the hepatorenal syndrome (second hit).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


