Primary clinical features
Gene(s)
Protein(s)
Disorder(s)
Cholangiopathy
CIRH1A
CIRH1A, cirhin; cirrhosis, autosomal recessive 1A
North American Indian childhood cirrhosis
CLDN1
CLDN1; claudin 1
Neonatal sclerosing cholangitis with ichthyosis and alopecia
Unknown
Unknown
Other neonatal sclerosing cholangitis
Hepatocellular cholestasis with cholangiopathy
ABCB4
ABCB4; ATP-binding cassette, subfamily B, member 4; also multidrug resistance protein 3 (MDR3)
High-γGT progressive familial intrahepatic cholestasis (PFIC); high-γGT intrahepatic cholestasis of pregnancy (ICP); small duct primary sclerosing cholangitis; hepatolithiasis and cholelithiasis with cholesterol sludge and stones
Hepatocellular cholestasis
BCS1L
BCS1L; BC1 (ubiquinol–cytochrome c reductase) synthesis-like
GRACILE syndrome; clinicopathologically distinct mitochondriopathy (Bjørnstad syndrome, Leigh syndrome)
ATP8B1
ATP8B1; ATPase, aminophospholipid transporter, class I, type 8B, member 1; also familial intrahepatic cholestasis 1 (FIC1)
Normal-γGT PFIC initially manifest as bland intracanalicular cholestasis (PFIC, type 1); “benign” recurrent intrahepatic cholestasis (BRIC; BRIC, type 1); normal-γGT ICP
ABCB11
ABCB11; ATP-binding cassette, subfamily B, member 11; also bile salt export pump (BSEP)
Normal-γGT PFIC initially manifest as cholestatic (neonatal) hepatitis (PFIC, type 2); BRIC, type 2; normal-γGT ICP
BAAT
BAAT; bile acid-CoA:amino acid N-acyltransferase (glycine N-choloyltransferase)
Normal-γGT PFIC, initially manifest as cholestatic (neonatal) hepatitis, not linked to ATP8B1 or ABCB11
TJP2
TJP2; tight-junction protein 2; also zonula occludens protein 2 (ZO2)
Normal-γGT PFIC, initially manifest as cholestatic (neonatal) hepatitis, not linked to ATP8B1 or ABCB11
VPS33B
VPS33B; vacuolar protein sorting 33 homolog B (yeast)
Arthrogryposis–renal dysfunction– cholestasis syndrome
VIPAS39
VIPAS39; VPS33B interacting protein, apical-basolateral polarity regulator, spe-39 homologue; also VIPAR
Arthrogryposis–renal dysfunction– cholestasis syndrome
MYO5B
MYO5B; myosin VB; also MYO5B
Microvillus inclusion disease
Unknown
Unknown
Normal- or high-γGT PFIC unlinked to other genes
Unknown
Unknown
Normal- or high-γGT ICP unlinked to other genes
Unknown
Unknown
Lymphedema–cholestasis syndrome
Unknown
Unknown
Autosomal-dominant BRIC
Hypercholanemia
SLC27A5
SLC27A5; solute carrier family 27 (fatty acid transporter), member 5; also bile acid-CoA ligase (BACL)
Anicteric or minimally icteric cholestasis of hypercholanemia (malabsorption); scant hepatitis
BAAT
BAAT; bile acid-CoA:amino acid N-acyltransferase (glycine N-choloyltransferase)
Anicteric or minimally icteric cholestasis of hypercholanemia (malabsorption); scant hepatitis
TJP2
TJP2; tight-junction protein 2; also zonula occludens protein 2 (ZO2)
Anicteric or minimally icteric cholestasis of hypercholanemia (malabsorption)
EPHX1
EPHX; epoxide hydrolase 1, microsomal (xenobiotic)
Hypercholanemia in isolation
Hyperbilirubinemia
UGT1A1
UGT1A1; UDP glucuronosyltransferase 1 family, polypeptide A1
Crigler–Najjar and Gilbert syndromes
ABCC2
ABCC2; ATP-binding cassette, sub- family C, member 2; also canalicular multiple organic anion transporter (cMOAT)
Dubin–Johnson syndrome
SLCO1B1/SLCO1B3
SLCO1B1; solute carrier organic anion transporter family, member 1B1; also organic anion transporting polypeptide 1B1 (OATP1B1) / SLCO1B3; solute carrier organic anion transporter family, member 1B3; also organic anion transporting polypeptide 1B3 (OATP1B3)
Rotor syndrome
11.2 ATP8B1 Disease
Severe ATP8B1 disease, the form of PFIC first identified (PFIC-1), was described among members of an extended Amish kindred. The surname of the progenitor couple was Byler, as was that of the children in the four consanguine sibships initially reported (Clayton et al. 1969). “Byler disease” thus came to be used indifferently for PFIC. Mild ATP8B1 disease, or BRIC (later “BRIC-1”), had been recognized a decade before (Summerskill and Walshe 1959). (A form of BRIC not linked to the ATP8B1 or ABCB11 loci also has been reported (Floreani et al. 2000).)
In both PFIC-1 and BRIC-1, biopsy at presentation finds predominantly canalicular and centrilobular cholestasis (Fig. 11.1a). The bile within canaliculi often seems unusually pale (gray-green rather than khaki, as in other cholestatic disorders), and the canaliculi may dilate so widely that the liver appears glandular. A few flecks of bile pigment can be found within hepatocytes and Kupffer cells. Proliferated bile ductules are not seen at portal-tract margins and bile plugs are not seen in canals of Hering or bile ducts. The interlobular bile ducts in PFIC-1, which comes to medical attention in infancy (BRIC-1 generally does not present so early in life), may be difficult to discern without immunohistochemical studies. These demonstrate small ducts, perhaps composed of only one or two cells (personal observations). This apparent hypoplasia, which may reflect subnormal bile flow, does not warrant the diagnosis of “bile duct paucity.” Aside from rosetting centered on bile plugs, the lobule exhibits remarkably little unrest in ATP8B1 disease. Hepatocytes are small and compact, and anisocytosis and anisokaryosis are slight. Although small hepatocytes with several nuclei are often encountered, pronounced cytoplasmic edema or frank giant-cell change is not. Rare necrotic individual hepatocytes can be found, but the reticulin network is not disrupted, with splaying apart of fibers, and the overall pattern is not one of giant-cell hepatitis. Hemopoietic elements are scant or absent.
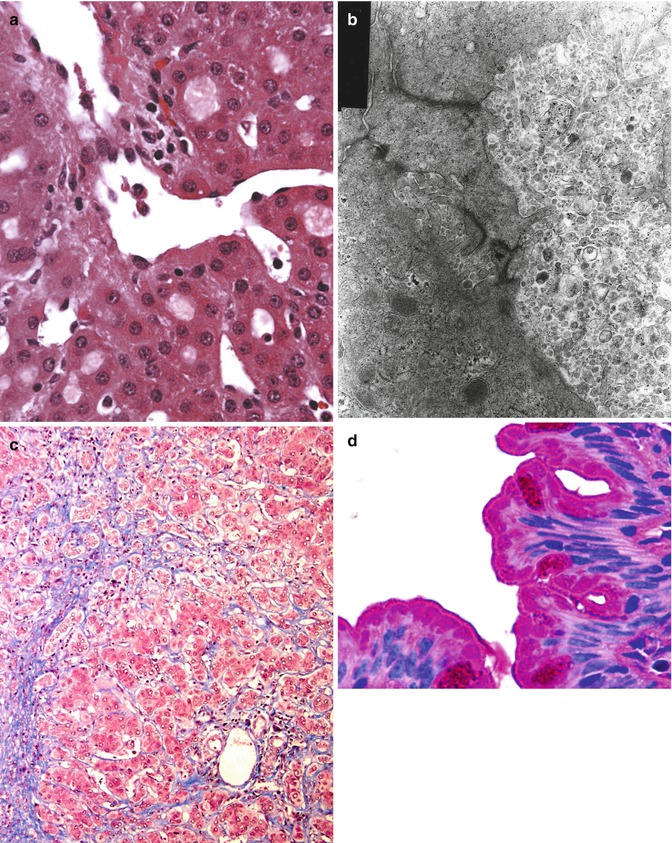
Fig. 11.1
(a) ATP8B1 disease; liver. Biopsy in early childhood; courtesy of Prof K Bove. Canaliculi are distended by pale bile; the centrilobular hepatocytes themselves are not edematous. Hematoxylin/eosin; original magnification, 400×. (b) ATP8B1 disease; liver. Biopsy at presentation in infancy; courtesy of Dr L Szönyi. Coarsely granular, loose “Byler bile” is characteristic in ATP8B1 disease. A distended canalicular lumen abuts a nearly normal one. Loss of microvilli and pericanalicular condensation of microfilaments are nonspecific. Osmium tetroxide/uranyl acetate/lead citrate; original magnification, 25,000×. (c) ATP8B1 disease; liver. Hepatectomy in childhood; courtesy of Dr C Bernard. Centrilobular region with cholangiolar metaplasia (compare portal tract in opposite corner). Masson trichrome; original magnification, 100×. (d) ATP8B1 disease; small bowel. Biopsy after LTX; courtesy of Dr C Bernard. Duodenal enterocytes have prominent subapical vacuoles distinct from mucin of goblet cells. The brush border is unremarkable. Periodic acid–Schiff; original magnification, 1,000×
Bile duct numbers can be evaluated easily using antibodies against cytokeratin (CK) 19. Immunostaining for CK7 also demonstrates bile ducts well; in addition, it may identify hepatocytes that mark diffusely for CK7, in both a cytoplasmic and a membranous pattern, lying individually or in small groups throughout the lobule. This is perhaps a nonspecific sequela of chronic cholestasis (Yabushita et al. 2001). Immunostaining for γGT in PFIC-1 finds reactivity at apices of cholangiocytes and along canaliculi of immediately periportal hepatocytes, but not along canaliculi deeper within the lobule (Paulusma et al. 2006). Immunohistochemical demonstration that ATP8B1/FIC1 (the protein encoded by ATP8B1) is present or absent (Eppens et al. 2001) is not yet a routine diagnostic option. Coarsely granular “Byler bile” (Fig. 11.1b) is found within canaliculi on ultrastructural study in both PFIC-1 and BRIC-1 (Bull et al. 1997; Phillips et al. 1987; Klomp et al. 2000; Chen et al. 2002). This feature is presumed to reflect abnormal bile composition, with shedding of unstable canalicular membrane into the lumen (Paulusma et al. 2006; Groen et al. 2008). Whether choleretics such as ursodeoxycholic acid alter the ultrastructural appearance of intracanalicular bile in ATP8B1 disease is not known. A “treatment holiday” before a scheduled biopsy thus may be prudent if transmission electron microscopy is intended. When interpreting ultrastructural findings, one should also bear in mind whether choleretics were being administered at biopsy. “Byler bile” cannot be identified with certainty in formalin-fixed material or material retrieved from paraffin-embedded samples (Bull et al. 1997), but it is demonstrable in snap-frozen tissue thawed in electron-microscopy fixative (personal observations). The ultrastructural appearance of post-hepatic bile in ATP8B1 disease is not described.
By definition, BRIC does not lead – even over years – to fibrosis within the liver (Luketic and Shiffman 1999). In PFIC-1, however, and in some patients whose clinical disorder is consistent with BRIC (episodic pruritus, icterus, and rarely cough (Chatila et al. 1996; Cissarek et al. 1998)), fibrosis develops. Such “BRIC” patients probably lie at points along the spectrum whose ends are PFIC-1 and strictly defined BRIC-1 (van Ooteghem et al. 2002). A fuller understanding of correlations between ATP8B1 mutations and clinical findings (Tygstrup et al. 1999; Klomp et al. 2004; Folmer et al. 2009) awaits definition of various “background” modifiers. The pattern of fibrosis in PFIC-1 or “intergrade” disorders is centrilobular as well as “biliary” (Knisely et al. 2001). Centrilobular fibrosis initially is perivenular and perisinusoidal, paralleling the distribution of cholestasis. Centrilobular involvement can be difficult to appreciate later in PFIC-1 because centrilobular hepatocytes may undergo metaplasia to a cholangiolar phenotype (Fig. 11.1c). The scarred centrilobular regions then mimic portal tracts that lack bile ducts. Portal–portal bridging fibrosis precedes portal-central bridging. Fibrosis is accompanied by only slight lymphocytic inflammation, and ductular metaplasia adjoining true portal tracts is not at all prominent. In late stages, canalicular cholestasis may become less pronounced than in material sampled earlier in the course of the disorder; hepatocyte disarray and necrosis remain very slight. Nodularity in a postnecrotic or postinflammatory pattern is not a feature. Hepatobiliary malignancy in genetically defined PFIC-1 has not been described. “Byler bile” persists into advanced PFIC-1, being found in second-decade hepatectomy materials (Bull et al. 1997). (The effects of biliary diversion (mechanical interruption of the enterohepatic circulation of bile acids) on “Byler bile” are not known, although the ultrastructural appearance of bile before and after diversion may vary (Kurbegov et al. 2003)).
As ATP8B1 is expressed at a wide range of sites (Bull et al. 1998), that ATP8B1 disease is a multisystem disorder is not surprising. Extrahepatic manifestations of ATP8B1 disease (some of which have been documented only in PFIC-1) include elevated sweat chloride concentrations (Lloyd-Still 1981; Bourke et al. 1996; Knisely et al. 1997), sensorineural deafness (Stapelbroek et al. 2009), delayed pubescence and growth even after liver transplantation (LTX) (Lykavieris et al. 2003; and personal observations), and loose stools (Bourke et al. 1996; Winklhofer-Roob et al. 1992). Disordered calcium homeostasis may extend beyond hypovitaminosis D (Nagasaka et al. 2004). Whether ATP8B1 disease increases risk of lung disease is a matter for speculation (Ray et al. 2010; Paulusma et al. 2011). Diarrhea tends to worsen after LTX (Lykavieris et al. 2003; Egawa et al. 2002) and may respond to bile salt chelation (Egawa et al. 2002). This interesting phenomenon may have to do with faulty gut regulation of uptake of bile acids and other substances (Oude Elferink et al. 2002; Chen et al. 2004). The allograft liver tends to develop a panlobular steatosis with variable steatohepatitis and fibrosis (Lykavieris et al. 2003; Miyagawa-Hayashino et al. 2009). Both diarrhea and hepatic steatosis after LTX may respond to biliary diversion (Nicastro et al. 2012), that is, to directing bile from the allograft liver away from the native small bowel.
Understanding of mechanisms underlying these aspects of ATP8B1 disease will perhaps come when the cellular function of ATP8B1 is more clearly understood. ATP8B1 is thought to act as an aminophospholipid “flippase” that maintains appropriate composition of bilayer lipid membranes (Bull et al. 1998; Ujhazy et al. 2001; Paulusma et al. 2008; Stone et al. 2012). However, it has flippase-independent functions, as yet obscure, necessary for normal expression of cell-apex proteins and for generation of normal microvilli (Verhulst et al. 2010). How ATP8B1 dysfunction results in the various clinical and morphologic features of PFIC-1 and BRIC is not established nor are the roles of a chaperone protein, CDC50A, in modulating intracellular transport of ATP8B1 and in permitting function of ATP8B1 at its destination understood (Paulusma et al. 2008; Folmer et al. 2012). Whether CDC50A disease may result in a phenocopy of ATP8B1 disease is an open question. While human disease owing to mutation in the orthologue ATP11C is not recognized, of note is that mutation in Atp11c, a gene encoding an enzyme structurally and functionally similar to ATP8B1, is associated in mice with an X-linked form of intrahepatic cholestasis (Siggs et al. 2011).
Intrahepatic cholestasis of pregnancy (ICP), which can be a low-γGT disorder, has been recorded in obligate heterozygotes for ATP8B1 disease (Clayton et al. 1969), but it is not a constant feature even among Amish women carrying the same ATP8B1 mutation (Bull et al. 1998) (personal observations). Low-γGT ICP not associated with demonstrable ATP8B1 mutation is well-recognized (Painter et al. 2005; Müllenbach et al. 2005), and ICP thus is to be regarded as genetically heterogeneous. One instance of successful pregnancy after LTX in PFIC-1 is reported; ICP was not observed (Cash et al. 2011).
11.3 ABCB11 Disease
Identification of mutations in ABCB11 as responsible for PFIC-2 (Strautnieks et al. 1998) rapidly followed demonstration that mutations in ATP8B1 underlie PFIC-1 (Bull et al. 1998). ABCB11 is the primary bile salt export protein at the hepatocellular canaliculus. Postnatal expression is restricted to the liver. Failure to secrete bile salts in ABCB11 disease explains both the lack of those salts in “mature bile” and – because of their cytotoxic activity at sites where they accumulate – the evidence of hepatocellular injury found on clinical–biochemistry analysis and in biopsy specimens.
Biopsy at presentation in severe ABCB11 disease finds giant-cell hepatitis. Hepatocellular and canalicular cholestasis; hepatocellular edema, giant-cell change, and necrosis; hemopoietic elements within the lobule; mononuclear-leukocyte inflammation (apart from sites of necrosis, where neutrophils gather); and fibrosis centered on portal tracts (Fig. 11.2a, b) characterize biopsy specimens at presentation (Bull et al. 1997; Davit-Spraul et al. 2010a; Chen et al. 2002; Pawlikowska et al. 2010; Evason et al. 2011). Serum concentrations of transaminase activities are more elevated than in ATP8B1 disease (Bull et al. 1997; Davit-Spraul et al. 2010a; Chen et al. 2002; Pawlikowska et al. 2010). The interlobular bile ducts, like those in PFIC-1, may be hypoplastic but are not lacking. Slightly proliferated ductules at portal-tract margins are usual; intraductular and intraductal cholestasis are not seen. As in ATP8B1 disease, hepatocytes express CK7 inappropriately. Canalicular membranes express γGT throughout the lobule (Paulusma et al. 2006); when available, immunohistochemical demonstration that ABCB11/BSEP (the protein encoded by ABCB11) is present or absent is useful (Davit-Spraul et al. 2010a; Evason et al. 2011; Jansen et al. 1999; Strautnieks et al. 2007) (Fig. 11.2c). Canalicular bile is dense, amorphous, or finely filamentous on ultrastructural study, but not coarsely granular (Bull et al. 1997; Chen et al. 2002; Evason et al. 2011).
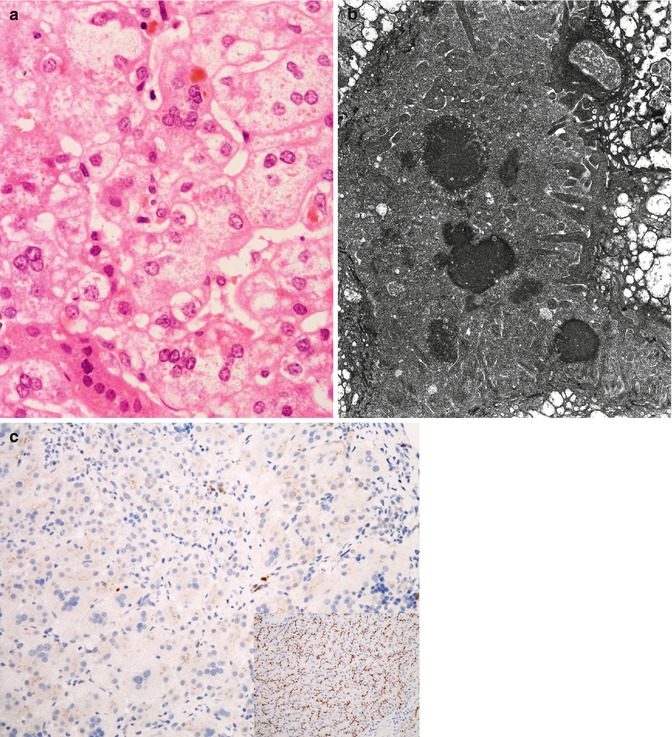
Fig. 11.2
(a) ABCB11 disease; liver. Biopsy at presentation in infancy. Giant-cell change and edema of hepatocytes with focal necrosis and bile pigment are usual in the lobule. Compare with Fig. 11.1a. Hematoxylin/eosin; original magnification, 400×. (b) ABCB11 disease; liver. Biopsy at presentation in infancy; courtesy of Dr D Zwick. Amorphous or filamentous bile, or electron-dense bile, is usual. Compare with Fig. 11.1b. Edema of hepatocyte cytoplasm is present. Osmium tetroxide/uranyl acetate/lead citrate; original magnification, 18,150×. (c) ABCB11 disease; liver biopsy. Staining for anti-BSEP antibody by immunohistochemistry is absent; inset shows staining pattern of normal control. Avidin–biotin–peroxidase with hematoxylin counterstain, 200×
As in PFIC-1, fibrosis worsens as the patient ages. It appears to progress more rapidly in ABCB11 disease than in ATP8B1 disease: Death or LTX supervenes in the first decade, as a rule, rather than in the first or second decade (the case with PFIC-1; personal observations). A tendency to giant-cell change and anisocytosis of hepatocytes may persist. Some centrilobular fibrosis is seen. It is less apparent than in PFIC-1, perhaps because it is obscured in PFIC-2 by septal and lobular chronic inflammation and disruption of architecture, with hepatocellular injury, regeneration, and nodular transformation (Davit-Spraul et al. 2010a; Pawlikowska et al. 2010; Evason et al. 2011). Biliary diversion may alter the natural history of ABCB11 disease (Arnell et al. 2010). Both hepatocellular carcinoma (Knisely et al. 2006) and cholangiocarcinoma (Scheimann et al. 2007) are reported in ABCB11 disease, as is a single instance of pancreatic ductal adenocarcinoma (Bass et al. 2010). BRIC associated with ABCB11 mutations has been reported and is termed BRIC-2 (van Mil et al. 2004). As with PFIC-1 and BRIC-1, intergrades between PFIC-2 and BRIC-2 are clinically and histopathologically apparent (Zellos et al. 2012). Although ABCB11 mutation predicted to abolish BSEP synthesis appears to confer increased risk of malignancy (Strautnieks et al. 2007), clinically less severe disease is not exempt (Sheridan et al. 2012). BSEP can transport species other than amidated bile acids (Hirano et al. 2005): Whether accumulation of bile acids, with disordered intracellular signaling as a result, or failure to eliminate other toxic compounds conduces to malignancy is not known.
Extrahepatic clinical and morphologic manifestations of ABCB11 disease are not recognized either before or after LTX. (A child with dysmorphism and hemizygosity for both an ABCB11 mutation and flanking genes, owing to microdeletion, is an exception (Strautnieks et al. 2001b).) LTX may appear curative rather than, as in PFIC-1, unmasking extrahepatic disease; the allograft liver in ABCB11 disease does not develop steatosis, and diarrhea after LTX is not encountered (personal observations). However, intrahepatic cholestasis after LTX for PFIC-2, repeatedly observed, that clinically and on microscopy mimics BRIC-2 or PFIC-2 (Keitel et al. 2009; Jara et al. 2009; Siebold et al. 2010; Maggiore et al. 2010) has been traced in some patients to formation of antibodies against BSEP that block transport function (Keitel et al. 2009; Jara et al. 2009; Dixon et al. 2009). While IgG can be demonstrated immunohistochemically at the bile canaliculus in such patients (Keitel et al. 2009; and personal observations), the incidence of canalicular IgG deposits after LTX for PFIC-2 is unstudied in patients whose BSEP function appears intact.
Like that of ATP8B1 disease, the contribution of ABCB11 disease to low-γGT ICP awaits full definition (Dixon et al. 2009), although some aspects are increasingly well understood. Estrogens alter activity of BSEP (Stieger et al. 2000). Shifts in intrahepatocytic location of BSEP between canalicular membrane and cytoplasm (Kipp et al. 2001) as well as in transcriptional regulation of ABCB11 (Ananthanarayanan et al. 2001) are implicated in some forms of cholestasis induced by shifts in hormonal milieu, infection, or drugs (Stieger and Geier 2011; Kubitz et al. 2011). ICP, or contraceptive-associated intrahepatic cholestasis, associated with ABCB11 mutation likely can be ascribed to such shifts.
Interactions among bile acids, estrogens, ATP8B1 expression and FIC1 function, and ABCB11 expression and BSEP function are remarkably complex, however, and defy summary here. FIC1, expressed constitutively in a manner independent of bile acids (Cebecauerová et al. 2012), induces BSEP expression via the central bile acid sensor farnesoid X receptor (FXR), encoded by NR1H4 (Chen et al. 2004; Alvarez et al. 2004; Frankenberg et al. 2008; Chen et al. 2010). This mechanism by which ATP8B1 mutation might lead through BSEP downregulation to cholestasis has been questioned (Cai et al. 2009). Although BSEP expression in ATP8B1 disease generally appears intact on immunohistochemical study (Davit-Spraul et al. 2010a; and personal observations), intact-appearing expression does not necessarily imply normal function, and decreased BSEP expression indeed is described in ATP8B1 disease (Alvarez et al. 2004; albeit personally observed only rarely). The substantial differences in histopathologic appearance of the liver between ATP8B1 disease and ABCB11 disease suggest regardless that cholestasis in ATP8B1 disease cannot be ascribed solely to retention of bile salts within hepatocytes. That is the postulated mechanism of injury in ABCB11 disease; if it were the mechanism of injury in ATP8B1 disease, then the hepatobiliary manifestations of the two disorders should, it seems reasonable to expect, resemble one another more closely than they do.
Variation in NR1H4 has been implicated in ICP (Van Mil et al. 2007), possibly via downregulation of BSEP expression (Ananthanarayanan et al. 2001). The extent to which altered FXR function contributes to other forms of intrahepatic cholestasis remains to be determined. NR1H4 disease has not been implicated in a small series of patients with intrahepatic cholestasis in whom ATP8B1 or ABCB11 lesions were not detectable (Davit-Spraul et al. 2012).
When assessing liver biopsy materials from a patient with suspected BRIC or PFIC, one must remember that while absence of a gene product can be identified immunohistochemically, insufficiency of expression of a gene product, or expression of a functionally deficient form of a gene product, cannot reliably be distinguished from wild-type expression. As with ICP in adulthood, carrier status for a mutant allele of ABCB11 may be associated with clinically manifest cholestasis in early infancy (Hermeziu et al. 2006; Liu et al. 2013). The same appears true for ATP8B1 (Jacquemin et al. 2010). This is likely multifactorial; the ontogenetically immature liver may cope poorly in several respects with transition to enteral feeding.
In older children and in adults, pubescence, drug exposure, or intercurrent illness can serve as a trigger for a bout of intrahepatic cholestasis. Less often encountered, and thus liable to be forgotten, is the unusual phenomenon of paraneoplastic intrahepatic cholestasis. (The histopathologist will do well to remind clinical colleagues of the entity.) Paraneoplastic intrahepatic cholestasis may be the result of bile duct injury and even loss, with high serum γGT activity. Less frequently, however, it may strongly resemble BRIC, with principally intracanalicular accumulations of bile pigment, only scant hepatocellular injury, and normal range serum γGT activity (Barta et al. 2006). The role of mutation in ATP8B1 or ABCB11 in this setting awaits study (Blackmore et al. 2013).
Histopathologic evaluation of intrahepatic cholestasis in the older patient is unlikely to yield findings that definitively implicate ABCB11 rather than ATP8B1 or vice versa. In the young patient with PFIC, the hepatobiliary system is at a steady state and the changes present are both pronounced and stable. In the older patient with intrahepatic cholestasis of recent onset, the disorder likely is in evolution, either worsening or improving, with a phenotype that shifts during observation: Ambiguity is to be expected. The temptation simply to describe the pattern of changes seen and to assign the nondiagnosis of “Cholestasis; clinical and genetic correlation required” can be strong. The chance exists, however, to do that correlation oneself while including the histopathologic findings and to define the implications for liver biopsy interpretation that inhere in a particular pharmacogenetic interaction. This means casting the genetic net widely, as the sketch above of interactions among FIC1, BSEP, FXR, and other species will have signaled. But it seems worth attempting.
11.4 Familial Hypercholanemia (FHCA; BAAT, EPHX1, SLC27A5, and TJP2 Disease)
Like PFIC and BRIC, FHCA is a designation encompassing several entities. It is likely soon no longer to be useful except as a clinical category, and a provisional one at that, to be used only until clinical-biochemistry and genetic analyses permit more precise classification.
A single instance is known of FHCA ascribed to deficiency of hepatocyte bile salt uptake from plasma (mediated by epoxide hydrolase, encoded by EPHX1). Compound heterozygosity for two EPHX1 mutations affecting promoter function was found in the proband, who exhibited hypercholanemia without jaundice or malabsorption (Zhu et al. 2003). Failure to thrive, the consequences of malabsorption of fat and fat-soluble vitamins, and pruritus without jaundice associated with elevated concentrations of serum bile salts – that is, anicteric rather than icteric cholestasis – are the usual clinical features of other disorders subsumed into FHCA, which was defined among Eastern Pennsylvania Amish and Mennonite children without demonstrable ATP8B1 disease, ABCB11 disease, or bile acid synthesis disorder. These children had various combinations of mutation in TJP2 and BAAT (Carlton et al. 2003). Other patients with FHCA of unknown genetic etiology have been described, however (Ujhazy et al. 2001), and even among the Eastern Pennsylvania Amish evidence exists for a third FHCA locus (Carlton et al. 2003).
TJP2 encodes a claudin, a protein around which tight junctions are assembled, and BAAT encodes an enzyme that mediates the second step in conjugating bile acids with taurine and glycine. Without normal BAAT function, bile acids in circulation are principally unconjugated. In Amish and Mennonite children, heterozygosity for the known BAAT mutation increases penetrance of homozygosity for the known TJP2 mutation. Both mutations are postulated to increase reflux of bile acids from bile into plasma, through “leaky” canalicular tight junctions or via increased back diffusion of unconjugated species; BAAT deficiency also may lead to presentation to BSEP of a substrate that is transported poorly. Clinical manifestations in this group of FHCA patients, unlike those in either ATP8B1 disease or ABCB11 disease, tend to improve with ursodeoxycholic acid therapy and tend to lessen as patients age (Shneider et al. 1997). Other children with BAAT disease owing to different mutations have since been described, of whom some have substantially more pronounced cholestasis (Hadžić et al. 2012; Setchell et al. 2013). Whether mutation in other cholestasis-associated genes was present in these children is moot. Other children with TJP2 mutation and severe cholestasis exist (Sambrotta et al. 2014).
Another disorder with features of FHCA is SLC27A5 disease. BACL, the product of SLC27A5, prepares bile acids for the action of BAAT. Two siblings with BACL deficiency are described (Chong et al. 2012). One, born prematurely and requiring parenteral alimentation, was jaundiced, without elevated serum γGT activity. Bile acids in circulation were principally unconjugated. She had no identifiable mutation in BAAT and was found to be homozygous for a predictably deleterious single-nucleotide substitution mutation in SLC27A5. She also was homozygous for a predictably deleterious single-nucleotide substitution mutation in ABCB11. Her younger sister was homozygous for the same SLC27A5 mutation and had principally unconjugated bile acids in circulation. She was not jaundiced. However, she was born at term and harbored only one copy of the ABCB11 mutation. That immaturity, parenteral alimentation, and homozygosity for ABCB11 mutation contributed to jaundice in the older sibling seems likely.
Histopathologic material from mutationally confirmed FHCA patients is scant. In the only children to date described with SLC27A5 mutation, mild “neonatal hepatitis” was seen in one who also was homozygous for an ABCB11 mutation. Immunostaining of a liver biopsy specimen from this child found expression of BACL and BSEP as well as of BAAT (Chong et al. 2012). In one infant with TJP2 disease compounded by heterozygosity for the known Amish/Mennonite BAAT mutation, although canalicular expression of γGT was not assessed, bland intracanalicular cholestasis (Fig. 11.3a) met light microscopic criteria for ATP8B1 disease (Maksimak et al. 1997). Ultrastructural study was not conducted. Genetic analysis did not find the G308V mutation in ATP8B1 that occurs among the Amish (Bull et al. 1998; Carlton et al. 2003). In another such patient who in early childhood came to biopsy (Fig. 11.3b) while receiving ursodeoxycholic acid, chronic hepatitis with minimal activity and mild portal-tract fibrosis were seen (Morton et al. 2000). Canalicular expression of γGT in this child was unremarkable (personal observations). Dilated and contorted canaliculi, containing amorphous loose material and bounded by hepatocyte tight junctions longer than in comparison specimens, were found on transmission electron microscopy (Carlton et al. 2003). The tight junctions were not “unzipped,” as noted in some instances of primary bile acid synthesis disorder (Bove et al. 2004). In infancy, light microscopy and ultrastructural study in subsequently described instances of BAAT disease without known TJP2 disease found changes of “neonatal hepatitis” with intralobular cholestasis (Hadžić et al. 2012; Setchell et al. 2013) that largely resolved on subsequent biopsy or changes of small duct cholangiopathy with fibrosis and intralobular cholestasis that progressed, requiring LTX (Setchell et al. 2013). When conducted, immunostaining found intact canalicular expression of BSEP and γGT and deficiency of BAAT expression (Hadžić et al. 2012; Setchell et al. 2013). Bile was amorphous rather than coarsely granular (Hadžić et al. 2012; Setchell et al. 2013).
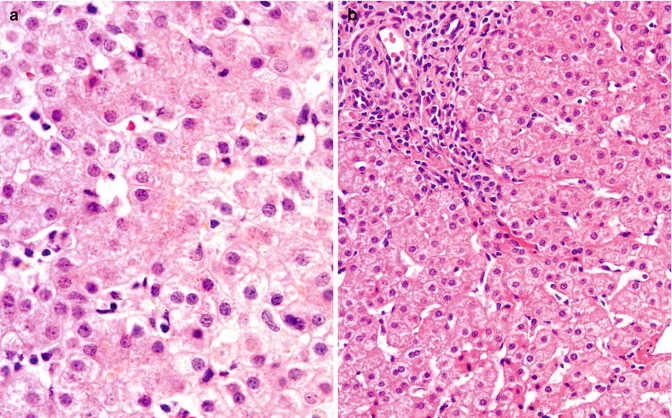
Fig. 11.3
(a) TJP2 disease with heterozygosity for BAAT mutation; liver. Biopsy at presentation in infancy; courtesy of Dr M Maksimak. Scant bile is seen within canaliculi; hepatocyte disarray is not a feature. Hematoxylin/eosin; original magnification, 400×. (b) TJP2 disease with heterozygosity for BAAT mutation; liver. Biopsy in childhood; courtesy of Dr D Belchis. Biopsy specimen taken while patient receiving ursodeoxycholic acid. Chronic hepatitis with mild activity and mild portal-tract fibrosis are found. Cholestasis is not a feature. Hematoxylin/eosin; original magnification, 200×
Extrahepatic manifestations in TJP2 disease may include slight facial dysmorphism with hypertelorism (Carlton et al. 2003; Morton et al. 2000). Whether this reflects claudin dysfunction is unknown. Systemic evaluation for tight-junction abnormalities (e.g., in bowel mucosa) has not been conducted in FHCA patients. ICP has not been recognized in obligate heterozygotes for TJP2, BAAT, or SLC27A5 mutations, and pregnancy has not occurred in known FHCA patients.
11.5 Arthrogryposis–Renal Dysfunction–Cholestasis (ARC) Syndrome
This autosomal-recessive multisystem disorder involves the central nervous system, kidney, liver, and skin. Its features include arthrogryposis, Fanconi-type aminoaciduria, low-γGT conjugated hyperbilirubinemia, and ichthyosis (Gissen et al. 2004; Nezelof et al. 1979; Hershkovitz et al. 2008). Affected persons generally die in infancy. Tubular and interstitial mineralization and anterior horn motor neuron degeneration are recognized (Gissen et al. 2004; Nezelof et al. 1979). Skin changes are associated with failure of lamellar granule secretion (Hershkovitz et al. 2008). Platelets may be abnormally large (Gissen et al. 2004; Lo et al. 2005) and functionally deficient (Lo et al. 2005). Mild to moderate hepatocellular and canalicular cholestasis, with some multinucleation of hepatocytes that falls short of frank giant-cell hepatitis (Fig. 11.4a), may be accompanied by accumulation of pigment like that seen in Dubin–Johnson syndrome (Gissen et al. 2004; Nezelof et al. 1979) without decreased canalicular ABCC2 expression (Fig. 11.4b). Bile ducts are unremarkable in personal experience, but paucity has been reported (Gissen et al. 2004); that not paucity but hypoplasia was present seems plausible. Dense, amorphous canalicular bile is seen on ultrastructural study (personal observations). Expression of γGT at the canalicular membrane is decreased, with apparent displacement of expression to cytoplasm and basolateral aspects (Gissen et al. 2004). The composition of bile in ARC syndrome has not been studied. An association of ARC syndrome with ICP has not been recognized.
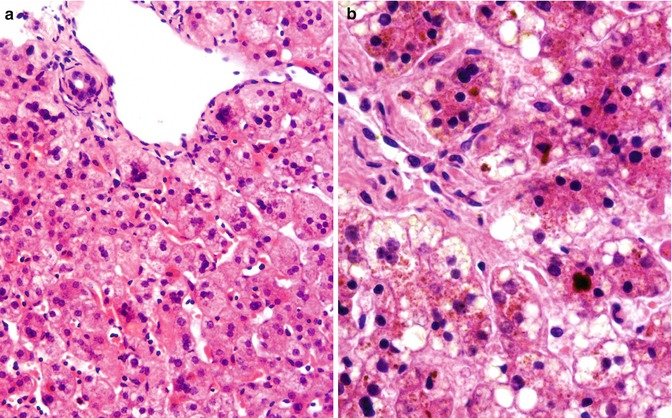
Fig. 11.4
(a) ARC syndrome; liver. Biopsy at presentation in infancy; courtesy of Dr P McKiernan. Slightly bile-tinged hepatocytes with scant intracanalicular cholestasis focally have several nuclei; florid giant-cell change and Dubin–Johnson-like pigment are not seen. Hematoxylin/eosin; original magnification 200×. (b) ARC syndrome; liver. Death in infancy; autopsy material, courtesy of Prof F Jaubert. Dubin–Johnson-like pigment and canalicular cholestasis can be seen. Hematoxylin/eosin; original magnification 400×
The genes mutated in patients with ARC syndrome, VPS33B and VIPAS39 (also VIPAR), encode proteins that interact with one another in the trafficking of cell membrane components to apical rather than to basolateral domains (Gissen et al. 2004; Cullinane et al. 2010). Mutations identified to date are from patients with illness manifest in early life and, correspondingly, most are predicted to ablate protein synthesis or function (Smith et al. 2012). In this respect, ARC syndrome thus may be considered analogous to PFIC, and in particular, as a multisystem disorder, to PFIC-1. As with PFIC-1, individual features of ARC syndrome vary in severity among patients. Arthrogryposis, for example, may be trivial or absent even though cholestasis is substantial (Bull et al. 2006), and not all patients with cholestasis manifest hepatocellular pigmentation (personal observations). As with ATP8B1 and ABCB11 in PFIC and BRIC, then, that dysfunction rather than absence of the proteins encoded by VPS33B and VIPAS39 may result in intermittent or late-onset clinical illness appears a reasonable expectation. Such illness has not been described, and the pharmacogenetics of VPS33B and VIPAS39 is unexplored territory. However, that either constitutive or environmentally induced dysfunction of these genes’ products may be implicated in some instances of intrahepatic cholestasis without full ARC syndrome is worth bearing in mind.
11.6 Microvillus Inclusion Disease (MVID; MYO5B Disease)
Like ATP8B1 disease and ARC syndrome, MVID is a multisystem disorder inherited in an autosomal-recessive pattern. Apical (adluminal) surfaces of various epithelia are imperfectly assembled or maintained in MVID, some components being inappropriately retained or endocytosed. These then lie subapically within epithelial cells (Phillips and Schmitz 1992; Reinshagen et al. 2002). Malabsorption and diarrhea are the primary manifestations, which have been treated by small bowel transplantation (Bunn et al. 2000; Halac et al. 2011). As with ARC syndrome, Fanconi-type renal dysfunction may be seen (Golachowska et al. 2012). MVID can involve the biliary tract (Rhoads et al. 1991). Mutation in MYO5B is associated with MVID (Müller et al. 2008). MYO5B, in conjunction with several Rab GTPases, subserves trafficking of proteins between the plasma membrane and intracellular vesicles; different regions of MYO5B interact with different Rab proteins (Roland et al. 2011). That some instances of MVID may be due to mutation in the genes encoding these partners of MYO5B rather than to mutation in MYO5B itself seems likely (Melendez et al. 2013), but such instances are not yet described. As with ARC syndrome and VPS33B/VIPAS39, contributions to hepatobiliary disease of mutation in MYO5B not associated with the MVID phenotype remain to be assessed.
Low-γGT intrahepatic cholestasis may develop in MVID patients after small bowel transplantation (Peters et al. 2001, Girard et al. 2014). On biopsy canalicular bile plugs are seen, with little hepatocellular disarray (Fig. 11.5a). One patient, whose ATP8B1 status remains to be determined, had “Byler bile” on ultrastructural study (Peters et al. 2001), but in others bile has been amorphous and either loose or dense. Subapical vacuoles like those in enterocytes have not been identified in hepatocytes (personal observations; and Girard et al. 2014). Expression of γGT at the canalicular membrane is decreased (personal observations; Fig. 11.5b). Concentrations of chenodeoxycholate salts in “mature bile” have been either normal or depleted (Peters et al. 2001, Girard et al. 2014). The prognosis of such cholestasis is unclear, as is its aetiology, although disordered handling of BSEP has been reported (Girard et al. 2014). Perhaps in MVID, as seems likely for ATP8B1 disease and for ARC syndrome, disorganisation of the hepatocyte apex underlies dysfunction of bile salt secretion.
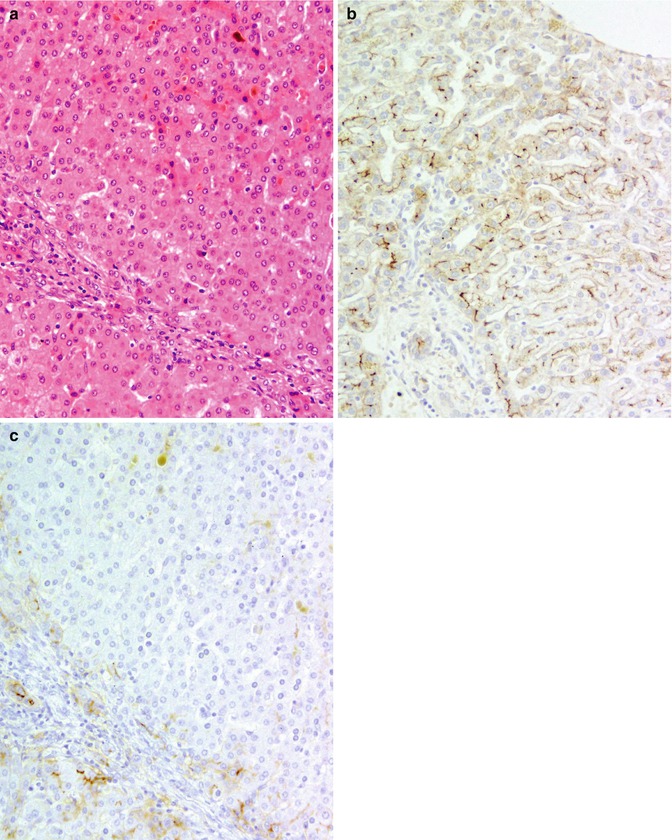
Fig. 11.5
(a) MVID; liver. Hepatectomy (combined hepatectomy and enterectomy); specimen courtesy of Dr F Lacaille. Predominantly intracanalicular cholestasis is found. Hematoxylin/eosin; original magnification 200×. (b) Normal liver. Expression of γGT (polyclonal rabbit antibody courtesy of Dr M Hanigan) is strong at cholangiocyte apices and along canalicular network. Avidin–biotin technique/hematoxylin; original magnification 200×). (c) MVID; liver. Specimen as in (a). Expression of γGT (source as in (b)) at the canalicular membrane is decreased within the lobule by comparison with normal liver (b). Avidin–biotin technique/hematoxylin; original magnification 200×
11.7 Primary Disorders of Bile Acid Synthesis
The synthetic route from cholesterol to cholic and chenodeoxycholic acids, the principal bile acids in man, has a number of steps and involves cytoplasmic, mitochondrial, and peroxisomal sites (Bove et al. 2004). Failure of cholesterol biosynthesis (Smith–Lemli–Opitz syndrome) (Honda et al. 1999; Rossi et al. 2005), failure of peroxisome assembly (Brosius and Gartner 2002), and single-gene mutations affecting individual pathway enzymes (frequently inferred from patterns of distribution of unusual bile acids) (Clayton et al. 1987; Zhao et al. 2012; Subramaniam et al. 2010; Kimura et al. 2012; Mizuochi et al. 2011; von Bahr et al. 2005; Setchell et al. 2003) thus may lead to disease (Table 11.2) (see also Chap. 13). Intrahepatocytic accumulations of bile acids are hypothesized to cause hepatocellular damage, thereby worsening hepatic function (Bove et al. 2004). In many instances, the clinical picture is one of low-γGT intrahepatic cholestasis without marked pruritus or elevations in serum bile acid concentrations, manifesting early in infancy as giant-cell hepatitis with fibrosis (Bove et al. 2004; Rossi et al. 2005; Clayton et al. 1987; Zhao et al. 2012; Subramaniam et al. 2010; Kimura et al. 2012; Mizuochi et al. 2011; von Bahr et al. 2005; Setchell et al. 2003). Presentation later in life, however, as a child with malabsorption in whom liver biopsy finds nonspecific cholestasis and scant inflammation (Akobeng et al. 1999), or in adulthood, as with cerebrotendinous xanthomatosis (Mizuochi et al. 2011), is not incompatible with a disorder of bile acid synthesis. Canalicular expression of γGT and of BSEP, where studied, has been unremarkable (personal observations). On ultrastructural study, canalicular bile is dense or amorphous and not coarsely granular. The interesting feature of disruption of canalicular tight junctions has been found in some patients (Bove et al. 2004).
Table 11.2
Primary and secondary disorders of bile acid synthesis (selected details in text)
Primary clinical features | Gene(s) | Protein(s) | Disorder(s) |
|---|---|---|---|
Giant-cell hepatitis in infancy | AKRID1 | AKRID1; aldo-keto reductase family 1, member D1 | Delta(4)-3-oxysteroid 5-beta-reductase deficiency |
HSD3B7 | HSD3B7; hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 7 | 3-Beta-hydroxy-delta-5-C27-steroid oxidoreductase deficiency; 3-beta-hydroxy- delta-5C-27-steroid dehydrogenase/isomerase deficiency | |
CYP7B1 | CYP7B1; cytochrome P450, family 7, subfamily B, polypeptide 1 | Oxysterol 7-alpha-hydroxylase deficiency | |
AMACR | AMACR; alpha-methylacyl-CoA racemase | Alpha-methylacyl-CoA racemase deficiency | |
Dysmorphism; failure to thrive | DHCR7 | DHCR7; 7-dehydrocholesterol reductase | Smith–Lemli–Opitz syndrome; 3-beta- hydroxysteroid delta 7-reductase deficiency |
Diarrhea (rarely giant-cell hepatitis with cholestasis) in childhood, with cataracts, xanthomata, and variable mental retardation | CYP27A1 | CYP27A1; cytochrome P450, family 27, subfamily A, polypeptide 1 | Cerebrotendinous xanthomatosis; sterol 27- hydroxylase deficiency |
Acute hepatic failure | Unknown | Unknown | “Neonatal hemochromatosis”; post-perinatal acute hepatic failure |
The principal disorder manifest in infancy from which bile acid synthesis disorders must be differentiated is ABCB11 disease. To demonstrate failure of BSEP expression immunohistochemically in patients with conjugated hyperbilirubinemia, serum γGT activity that is low or normal range and, at liver biopsy, “neonatal hepatitis” can identify ABCB11 disease, when present, in all but the small subset of patients who express dysfunctional BSEP (Strautnieks et al. 2007). Mass spectrometry of urine distinguishes patients with ABCB11 disease from patients with disorders of bile acid synthesis in that urine of the former lacks salts of bile acids synthesized by secondary pathways. Speciation of urine bile acids, alcohols, and salts also permits identification of individual defects in bile acid synthesis and, as a class, of bile acid conjugation defects (Chong et al. 2012). Immunostaining of liver biopsy materials can be useful in distinguishing between bile acid conjugation defects (Hadžić et al. 2012; Setchell et al. 2013). Its value in demonstrating individual bile acid synthesis defects is not established.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


