Disease
Proposed cause
Selective IgA deficiency
Impaired IgA synthesis; molecular defect unknown in most cases; mutation in TNFRSF13B in ~5 %
Common variable immunodeficiency
Impaired B cell maturation; molecular defect unknown in most cases; mutation in TNFRSF13B in ~10–15 %
X-linked agammaglobulinemia
Mutation in BTK results in absence of Btk in B cells
Hyper-IgM syndrome
Mutation in CD40L, leading to absence of CD40 ligand on T cells
Hyper-IgE syndrome
Mutation in STAT3 in autosomal dominant form; mutation in DOCK8 in autosomal recessive form
Severe combined immunodeficiency
Multiple defects; most common is due to defect in common γ chain; others include adenosine deaminase deficiency, purine nucleoside deficiency, and T cell receptor deficiencies
Omenn’s syndrome
Hypomorphic missense mutation in RAG1/2, IL-7Rα,RMRP, ADA, DNA ligase IV, Artemis, γc
DiGeorge syndrome
Thymic hypoplasia; microdeletion in 22q11.2
Chronic mucocutaneous candidiasis
Heterogeneous disorder; mutation in AIRE gene in some; mutation in STAT1 in autosomal dominant form
Wiskott-Aldrich syndrome
Mutation in WASP; gene involved in cell trafficking and motility
Chronic granulomatous disease
Mutation in gene for component of NADPH oxidase; CYBB mutation in X-linked form; mutation in CYBA, NCF1, NCF2, or NCF4 in autosomal recessive forms
Immune dysregulation, polyendocrinopathy, and enteropathy, X-linked (IPEX)
Mutation in FOXP3
Table 5.2
Gastrointestinal and hepatic infections in primary immunodeficiency
Disease | Gastrointestinal infections | Hepatic infections |
|---|---|---|
Selective IgA deficiency | Giardia intestinalis; strongyloidiasis | |
Common variable immunodeficiency | Giardia intestinalis; Cryptosporidium; cytomegalovirus | Hepatitis C |
X-linked agammaglobulinemia | Giardia intestinalis; Cryptosporidium | Hepatitis C; other viruses reported include coxsackie, ECHO |
Salmonella, Campylobacter | ||
Rotavirus, coxsackie virus, poliovirus | ||
Hyper-IgM syndrome | Giardia intestinalis, Cryptosporidium, Entamoeba histolytica, Salmonella, Histoplasma capsulatum | Biliary cryptosporidial infections; hepatitis B, hepatitis C, ECHO, histoplasmosis, Bartonella |
Hyper-immunoglobulin E syndrome | Esophageal cryptococcal infection; GI mycobacterial infection; ileocecal histoplasmosis | |
Severe combined immunodeficiency | Candida; Salmonella and other bacterial pathogens; cytomegalovirus, rotavirus, Epstein-Barr virus | Adenovirus; cytomegalovirus; hepatitis B; hepatitis C |
DiGeorge syndrome | Candida | |
Chronic mucocutaneous candidiasis | Candida; Histoplasma capsulatum | |
Chronic granulomatous disease | Hepatic abscess due to staphylococcal infection | |
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) | Cytomegalovirus |
Table 5.3
Inflammatory gastrointestinal and hepatic lesions in primary immunodeficiency
Disease | GI manifestation | Hepatic manifestation |
|---|---|---|
IgA deficiency | Celiac disease | Autoimmune hepatitis |
Food allergies | ||
Crohn’s disease-like lesion | ||
Nodular lymphoid hyperplasia | ||
Common variable immunodeficiency | Multifocal atrophic gastritis +/− intestinal metaplasia | Nodular regenerative hyperplasia |
Villous atrophy | Primary biliary cirrhosis (adults) | |
Nodular lymphoid hyperplasia | Autoimmune hepatitis | |
Crohn’s disease-like lesion | Hepatic granulomas | |
Granulomatous enteropathy | Idiopathic chronic hepatitis | |
Colitis (ulcerative colitis-like; lymphocytic colitis) | ||
X-linked agammaglobulinemia | Crohn’s disease-like lesion | Hepatic veno-occlusive disease |
Perianal fistula and perianal abscess | ||
Hyper-IgM syndrome | Nodular lymphoid hyperplasia | Sclerosing cholangitis |
Oral and perianal ulcers | ||
Severe combined immunodeficiency | Graft-versus-host disease-like lesion, small bowel, and colon | Graft-versus-host disease |
Esophageal reflux | Veno-occlusive disease | |
Omenn’s syndrome | Graft-versus-host disease-like lesion, small bowel, and colon | |
Chronic Mucocutaneous Candidiasis | Atrophic gastritis | |
Autoimmune polyglandular syndrome, type 1 | Autoimmune hepatitis | |
Wiskott-Aldrich syndrome | Crohn’s disease-like lesion involving colon | |
Chronic granulomatous disease | Esophageal and gastric outlet obstruction; Crohn’s disease-like lesion in small bowel; colitis (ulcerative colitis-like and Crohn’s-like) | Nodular regenerative hyperplasia |
Pigmented macrophages | Granulomas | |
Small vessel vasculopathy | ||
Sclerosing cholangitis | ||
Idiopathic chronic hepatitis | ||
Drug-induced liver injury | ||
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) | Autoimmune enteropathy | Autoimmune hepatitis |
Table 5.4
Malignancies involving the gastrointestinal tract and hepatobiliary system in primary immunodeficiency
Disease | Gastrointestinal malignancy | Hepatobiliary malignancy |
|---|---|---|
IgA deficiency | Lymphoma | |
Gastric adenocarcinoma | ||
Common variable immunodeficiency | Gastric adenocarcinoma | Hepatocellular carcinoma (adults) |
B cell lymphoma, involving small bowel | ||
Adenocarcinoma of the colon, +/− neuroendocrine features | ||
X-linked agammaglobulinemia | Non-Hodgkin’s lymphoma | |
Gastric adenocarcinoma | ||
Colorectal adenocarcinoma | ||
Hyper-IgM syndrome | Plasma cell proliferation | Hepatocellular carcinoma |
Colorectal carcinoma | Biliary carcinomas | |
High-grade neuroendocrine | ||
carcinomas of GI tract and biliary tree | ||
Severe combined immunodeficiency | GI Lymphoma | Lymphoma |
EBV-associated smooth muscle tumor | Posttransplant lymphoproliferative disorder | |
EBV-associated smooth muscle tumor | ||
Wiskott-Aldrich syndrome | GI lymphoma | |
Ataxia-telangiectasia | Gastric adenocarcinoma |
Hepatic pathology associated with immune disorders is often iatrogenic and related to treatment of the underlying condition. For instance, many patients receiving intravenous immunoglobulin in the 1990s and earlier were infected with hepatitis C virus from contaminated pooled products. Similarly, highly active antiretroviral therapy (HAART) regimens for treatment of human immunodeficiency virus (HIV) infection have virtually eliminated many of the unusual hepatic infections seen in the past but may result in therapy-related hepatic injury. Largely idiopathic conditions such as nodular regenerative hyperplasia are relatively common in some primary immunodeficiencies, and unusual hepatic tumors may be encountered in some cases.
5.1.1 Humoral Immunodeficiencies
5.1.1.1 Selective IgA Deficiency
Selective IgA deficiency, defined as a serum IgA concentration of less than 50 μg/ml, is the most common primary immunodeficiency (Yel 2010), occurring in 1 in 600 of Northern European ancestry (Burrows and Cooper 1997). The disorder is 20 times more common in whites than in African Americans (Burrows and Cooper 1997), and the incidence is lowest among Asian populations (Feng 1992; Kanoh et al. 1986).
A maturation defect of B cells to produce IgA is commonly observed in IgA-deficient patients. Mutations in the gene TNFRSF13B have been identified and associated with IgA deficiency (Salzer et al. 2005; Castigli et al. 2005; Rachid et al. 2006). This gene encodes a member of the tumor necrosis factor-receptor superfamily, the transmembrane activator and calcium-modulator and cyclophilin-ligand interactor (TACI), which mediates isotype switching in B lymphocytes. Alterations in the gene may cause defective terminal maturation of B cells into IgA-secreting plasma cells (Sakurai et al. 2007). The same TACI mutation may be seen in some patients with common variable immune deficiency (CVID). Defects in antibody production in patients with IgA deficiency might represent a continuum with those seen in CVID, and 20–30 % of IgA-deficient patients will also have deficits in IgG subclasses, especially IgG2. IgA-deficient patients with IgG2 deficiency are prone to infection.
Two subclasses of IgA have been described: subclass IgA1 in a monomeric form, mainly found in the blood circulation, and subclass IgA2 (secretory IgA) in a dimeric form, the dominant immunoglobulin in mucosal secretions. Secretory IgA has prime importance in protecting the mucosal surface against microbial infections and excluding antigens entering through the mucosal route (Yel 2010). Deficiency in IgA, therefore, may cause recurrent infection of the respiratory and GI tracts, allergic disorders, and autoimmune manifestations. However, most IgA-deficient patients are asymptomatic, which may be attributed to compensatory increase in secretory IgM in these patients.
5.1.1.1.1 Clinical Manifestations
Clinical manifestations of IgA deficiency range from no symptoms to recurrent infections, generally involving mucosal surfaces, autoimmune disorders, allergic diseases, and malignancy (Yel 2010). Symptoms are typically milder than seen with CVID, although recurrent upper and lower respiratory tract infections are common in both disorders. Similarly, GI manifestations of IgA deficiency are the same as those associated with CVID. Infections are less common than might be expected, possibly due to compensation for lack of mucosal IgA by transport of IgM across the mucosa into the gut lumen, but include acute diarrheal illnesses due to bacterial enterocolitis, and chronic diarrhea and malabsorption due to persistent Giardia lamblia infection. Chronic strongyloidiasis has also been reported (Leung et al. 1995). Susceptibility to insulin-dependent diabetes mellitus and celiac disease may be inherited together with IgA deficiency; all three conditions are linked to particular major histocompatibility haplotypes and probably represent genetically linked susceptibilities in certain populations. The prevalence of celiac disease, the most common noninfectious GI complication of IgA deficiency, is 7.7 % in children with IgA deficiency, compared to 1 in 500 in the general population (Meini et al. 1996). IgG but not IgA anti-tissue transglutaminase should be used as a screening tool in this population. A sprue-like illness characterized by chronic diarrhea with villous atrophy that does not respond to a gluten-free diet may be seen in IgA deficiency (Joo et al. 2009), as in CVID. Some patients may develop malabsorption due to nodular lymphoid hyperplasia (NLH) (Joo et al. 2009; Postgate et al. 2009). Pernicious anemia is seen more commonly in IgA deficiency than in CVID. As with other B cell disorders, the incidence of Crohn’s disease and gastric adenocarcinoma appears to be increased in IgA deficiency (Lai Ping So and Mayer 1997).
5.1.1.1.2 Pathology of IgA Deficiency
Persistent Giardia infection can lead to villous flattening secondary to trophozoites effacing and damaging the mucosa (Agarwal and Mayer 2010). The morphology of celiac disease occurring in the setting of IgA deficiency is similar to that seen in immunocompetent patients, but IgA-producing plasma cells are absent. Gastric biopsy from IgA-deficient patients with pernicious anemia shows chronic atrophic autoimmune gastritis-type morphology, with loss of parietal cells and a variable mononuclear inflammatory infiltrate. Diffuse NLH has been reported in isolated case reports in association with IgA deficiency (Postgate et al. 2009). NLH presents as multiple, large lymphoid aggregates found in the lamina propria and/or the submucosa of the small intestine, occasionally in the large intestine and stomach, sometimes associated with mucosal flattening causing malabsorption and diarrhea (Postgate et al. 2009). The lymphoid aggregates are predominantly composed of IgM-bearing cells, suggestive of compensatory IgM production.
Partial IgA deficiency has been linked to autoimmune hepatitis in children and is reported to be more commonly in LKM-1-positive patients than in the setting of ANA/SMA positivity (Gregorio et al. 1997), but specific liver diseases are not otherwise reported for this disorder.
5.1.1.1.3 Treatment
Asymptomatic IgA-deficient patients do not need any treatment. If these patients present with GI or liver diseases, they should be treated the same as a patient without this immunodeficiency. Nodular lymphoid hyperplasia in IgA-deficient patients may respond to oral steroids.
5.1.1.2 Common Variable Immunodeficiency
Common variable immunodeficiency (CVID), though not a common disorder, is probably the most common symptomatic primary immunodeficiency. It is characterized immunologically by hypogammaglobulinemia involving multiple antibody classes and a diminished ability to produce antibody vaccine and infection. T cell abnormalities are common; below-normal proliferative responses to mitogens are found in 40 % of patients, and 20 % have a relative lack of CD4 T cells (Cunningham-Rundles et al. 1999). The common abnormality shared by IgA deficiency and CVID is failure of terminal maturation of B lymphocytes into plasma cells producing various Ig subtypes. A primary B cell defect is favored in many patients, but in others defective antigen responsiveness in T helper cells may be the underlying basis for the disorder (Conley and Cooper 1998).
A genetic basis has long been suspected, based on the observation that familial inheritance of CVID occurs in 20 % of cases (Hammarstrom et al. 2000) and that CVID and IgA deficiency tend to occur in the same family; individual family members may gradually convert from one disorder to the other. In multiple case families, CVID is often present in parents, with IgA deficiency in the offspring, consistent with the hypothesis that CVID may develop later in life as a more severe manifestation of a common defect involving immunoglobulin class switching. Studies of isotype switching led to the discovery that the gene TNFRSF13B, which encodes TACI, is mutated in approximately 10–15 % of patients with CVID and 5 % of patients with IgA deficiency (Castigli and Geha 2006). TACI mediates isotype switching and induces apoptosis in B cells, and this loss of function may be the basis for the susceptibility of patients with TACI mutations for autoimmune and lymphoproliferative disorders (Castigli and Geha 2006). Molecular defects resulting in CVID have also been described in the costimulatory molecule inducible costimulator (ICOS), CD19-B cell receptor complex (CD19, CD21, and CD81), and B cell differentiation antigen, CD20 (Yong et al. 2011).
5.1.1.2.1 Clinical Manifestations
Clinical and immunological features of CVID are heterogeneous, but most patients present with recurrent bacterial infections, usually involving the upper and lower respiratory tracts, but other infections including those of the GI system can also occur. Patients may present with CVID at any age, ranging from infancy to late adult life, and males and females are equally affected. Autoimmune manifestations such as thyroid dysfunction, pernicious anemia, autoimmune hemolytic anemia, autoimmune thrombocytopenia, and rheumatoid arthritis are common, and granulomatous involvement of skin and visceral organs mimicking sarcoidosis may be seen (Cunningham-Rundles et al. 1999; Salzer and Grimbacher 2006). Patients with CVID may also present with noninfectious GI diseases, lymphoid proliferation, and an increased risk of malignancy. Chronic GI disorders resulting in diarrhea, malabsorption, and weight loss occur in about 20 % of CVID patients.
Chronic infection with Giardia intestinalis is a common problem in patients with CVID, although the prevalence of Giardia infections in this population appears to be decreasing (Lai Ping So and Mayer 1997). Giardia infection may be asymptomatic, but in some cases, malabsorption, steatorrhea, and villous abnormalities can be reversed if Giardia is eradicated. Other GI infections are less common in CVID. Cryptosporidiosis is occasionally found. The prevalence of common bacterial intestinal infections such as Salmonella and Campylobacter infection does not appear to be increased. Although prolonged antibiotic use is common in these patients, an increase in pseudomembranous colitis is not reported (Lai Ping So and Mayer 1997). On occasion, other viral and fungal organisms infect the GI tract in CVID patients, but such infections are less common in CVID than in acquired immunodeficiency syndrome (AIDS).
Helicobacter pylori infection is common in patients with CVID. Atrophic gastritis resulting in pernicious anemia may occur in the absence of demonstrable anti-parietal cell antibodies in these patients (Malamut et al. 2010). In the stomach, atrophic gastritis may develop at a very young age in patients with CVID and has been reported in a 6-year-old who developed multifocal gastric adenocarcinoma at age 11 (Conley et al. 1988). Adults with CVID are also at increased risk for gastric adenocarcinoma (Salzer and Grimbacher 2006). It has been estimated that patients with CVID have a 47-fold increase in gastric carcinoma compared to the general population of Great Britain (Cunningham-Rundles et al. 1987), and 5–10 % of CVID patients ultimately develop gastric carcinoma, usually many years after the onset of hypogammaglobulinemia.
In the small intestine, a sprue-like lesion occurs in some patients with CVID and is associated with severe malabsorption, often requiring parenteral nutrition (Malamut et al. 2010). This lesion usually does not respond to a gluten-free diet, although an elemental diet may be beneficial. Some patients with CVID develop chronic inflammatory processes involving small or large bowel and clinically similar to inflammatory bowel disease (Washington et al. 1996; Daniels et al. 2007). Granulomatous enteropathy has also been reported in patients with CVID and may be associated with protracted diarrhea unresponsive to antibiotic therapy (Ardeniz and Cunningham-Rundles 2009).
NLH is found in the small intestine, large intestine, or both in up to 60 % of patients with CVID (Malamut et al. 2010; Daniels et al. 2007; Luzi et al. 2003). NLH is not considered a malignant disorder. However, malignant lymphomas of the GI tract in patients with immunodeficiencies often arise in a background of NLH, and clonal immunoglobulin gene rearrangement has been demonstrated in nodular lymphoid hyperplasia in the GI tract of a child with CVID (Laszewski et al. 1990). Consistent with these observations, the most common malignancy in CVID is non-Hodgkin’s lymphoma, affecting approximately 8 % of patients (Cunningham-Rundles et al. 1999); these lymphomas often originate in extranodal sites, with small bowel the most common GI site. Most of these lymphomas are of B cell origin and include diffuse large B cell lymphoma and follicular lymphoma (Salzer and Grimbacher 2006).
Adenocarcinoma of the colon is reported in young patients with CVID. Small cell neuroendocrine carcinoma of the cecum has been reported in a 16-year-old boy, who died of liver metastases 5 months after diagnosis (de Bruin et al. 1993). In another case, nine adenocarcinomas and 20 adenomas were present synchronously in the colon of a 22-year-old man with CVID (Adachi et al. 1992).
5.1.1.2.2 Pathology of CVID
Stomach. In the stomach, a nonspecific increase in lamina propria lymphocytes is seen in some patients with CVID; increased apoptosis of gastric epithelial cells is present in some cases (Washington et al. 1996) (Fig. 5.1). In a study of gastric biopsies from 34 patients with CVID and dyspepsia, 41 % of patients were infected with Helicobacter pylori. All H. pylori-positive patients and 20 % of H. pylori-negative patients had chronic gastritis, and 50 % of those infected with H. pylori had multifocal atrophic gastritis. Ten percent of H. pylori-negative patients had multifocal atrophic gastritis (Zullo et al. 1999). Similarly, Daniels and colleagues report autoimmune atrophic gastritis, showing increased lamina propria lymphocytes, parietal cell loss, and intestinal metaplasia on biopsy of the gastric body (Fig. 5.2), in a small number of patients (Daniels et al. 2007). Plasma cells are not present. Granulomas may be found on gastric biopsy.
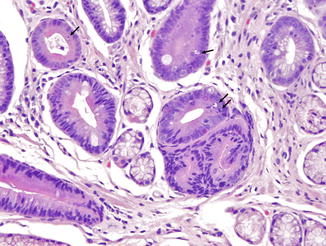
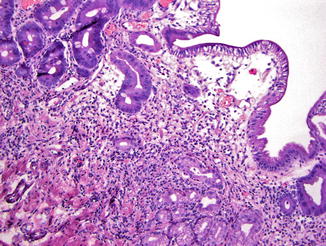
Fig. 5.1
Common variable immunodeficiency. Increased apoptotic bodies (arrows) in the gland can be seen in the gastric mucosa
Fig. 5.2
Common variable immunodeficiency. Autoimmune atrophic gastritis in CVID patients is characterized by increased lamina propria inflammation, loss of parietal cells, pyloric gland metaplasia, and intestinal metaplasia. Plasma cells are not present
Small Intestine. Small bowel mucosal abnormalities in giardiasis include villous blunting, increased intraepithelial lymphocytes, and nodular lymphoid hyperplasia. The trophozoite form of the organism can be identified among intestinal villi on small bowel biopsy (Fig. 5.3). Cytomegalovirus infection involving the esophagus, stomach, jejunum, and ileocecal area and resulting in multiple ulcers and obstructing strictures has been reported in a patient with CVID (Tahan et al. 2000).
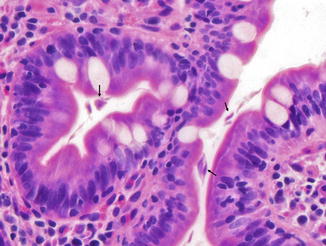
Fig. 5.3
Common variable immunodeficiency. Giardia trophozoites (arrows) are present between the duodenal villi
Increased intraepithelial lymphocytosis with villous atrophy can been seen in 31–60 % of patients with CVID (Washington et al. 1996; Daniels et al. 2007; Luzi et al. 2003; Khodadad et al. 2007) (Fig. 5.4). Villous atrophy associated with CVID generally lacks the degree of crypt hyperplasia seen in celiac disease but may be indistinguishable on biopsy. In general the lamina propria inflammatory infiltrate is not as prominent as in celiac disease, and enterocyte maturation is normal, with preservation of the brush border (Washington et al. 1996). Plasma cells are absent or found only in very small numbers in the lamina propria in CVID. Surface intraepithelial lymphocytes are often markedly increased, even in the absence of villous atrophy (Fig. 5.4b). In some cases, an increase in apoptotic bodies is found in crypt epithelial cells.
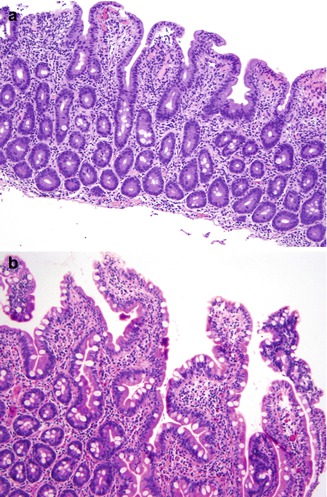
Fig. 5.4
Common variable immunodeficiency. A sprue-like lesion in the duodenum shows villous atrophy and prominent intraepithelial lymphocytes (a). Increased intraepithelial lymphocytes can be seen in absence of villous atrophy (b)
Poorly formed non-necrotizing granulomas are found in the lamina propria in approximately 10 % of small intestinal biopsies (Mike et al. 1991). Some patients with CVID can develop chronic inflammatory processes involving small or large bowel. In some patients, the small bowel is the primary site of involvement and the lesions resemble Crohn’s disease, with transmural inflammation and small bowel obstruction. Granulomas are generally not present in these Crohn’s disease-like disorders (Hermaszewski and Webster 1993).
NLH in the GI tract is characterized by multiple discrete hyperplastic lymphoid nodules in the lamina propria and submucosa of the small intestine (Fig. 5.5), less commonly large intestine, or both, and is probably a result of chronic antigenic stimulation (Agarwal and Mayer 2010). The germinal centers of the follicles are composed of proliferating B cells with scattered tingible body macrophages; the mantle zones contain mature and immature B cells, and the extramantle zones contain a mixture of cell types including B and T cells and macrophages (Washington et al. 1996). In contrast to NLH in non-immunodeficient patients, plasma cells are absent in the extramantle zones in CVID patients.
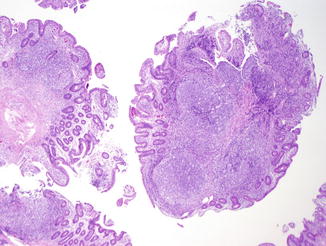
Fig. 5.5
Common variable immunodeficiency. There are multiple discrete hyperplastic lymphoid nodules in the lamina propria and submucosa of the small intestine
Large Bowel. The colitis occurring in CVID is variable in morphology. In some patients the inflammatory process is limited to the colon and clinically mimics ulcerative colitis. Mucosal architectural distortion with crypt destruction is present, although crypt distortion is less pronounced than in most cases of ulcerative colitis, with less crypt branching (Fig. 5.6a). Neutrophils are present in the lamina propria and crypt epithelium (Fig. 5.6b). In contrast to ulcerative colitis, plasma cells are not present in the lamina propria in CVID-associated colitis. In some cases, the crypt destruction and mucosal distortion is accompanied by increased apoptosis, and the histology is similar to colonic graft versus host disease (Washington et al. 1996). Milder cases of colitis in CVID may resemble lymphocytic colitis (Fig. 5.7), characterized by increased intraepithelial lymphocytes and minimal mucosal distortion (Daniels et al. 2007).
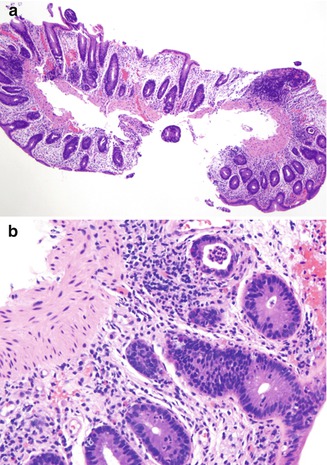
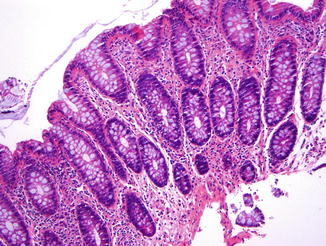
Fig. 5.6
Common variable immunodeficiency. Ulcerative colitis-like lesion shows mild architectural distortion (a). Cryptitis and crypt abscess can be seen (b). No plasma cells are present in the lamina propria
Fig. 5.7
Common variable immunodeficiency. Lymphocytic colitis pattern in CVID shows markedly increased intraepithelial lymphocytes. However, the lamina propria inflammation is not as prominent as typical lymphocytic colitis
5.1.1.2.3 Liver Disease in CVID
Clinical Manifestations. Persistent elevation in liver tests is reported in up to 20 % of patients with CVID. Hepatosplenomegaly is a common presentation and can fluctuate over a lifetime. Autoimmune liver diseases associated with CVID include primary biliary cirrhosis (seen in 5 % of adults with CVID in one series (Cunningham-Rundles et al. 1999)) and autoimmune hepatitis. Patients with CVID may also present with portal hypertension, which is generally a result of nodular regenerative hyperplasia (NRH), associated with autoimmune diseases and azathioprine therapy in one series (Malamut et al. 2008) and polyclonal lymphoid proliferations and enteropathy in another (Ward et al. 2008). NRH does not appear to be associated with age of onset of CVID or with length of IVIG therapy (Ward et al. 2008). Because treatment of CVID with IVIG entails risk of viral infection from a transfused blood-derived product, some patients are exposed to potential hepatotropic pathogens such as hepatitis C virus, although risk of viral infection is decreased with modern IVIG processing methods.
Liver Pathology. The most commonly reported patterns of injury in liver in CVID patients are nodular regenerative hyperplasia, chronic hepatitis, and hepatic granulomas. Hepatic malignancies, including hepatocellular carcinoma arising in the absence of preexisting liver disease, have been described in adults with CVID (Gandhi et al. 2010) but have not yet been reported in pediatric patients.
Nodular regenerative hyperplasia is one of the most common findings on liver biopsy in patients with CVID, but it may be difficult to diagnose on needle biopsy. The prevalence in biopsies is reported in the range of 80–87 % (Malamut et al. 2008; Ward et al. 2008). NRH was overlooked on initial evaluation in more than half the biopsies in one series (Malamut et al. 2008). Grossly, the liver in NRH appears nodular, and the capsule may have a subtle pigskin texture; the gross impression of cirrhosis and the finding of relatively minor fibrosis on histologic examination should prompt consideration of NRH. Histologically, nodularity of the parenchyma without bridging fibrosis is seen, and alternating areas of thickened and atrophic liver cell plates are appreciated. Compression of sinusoids may produce congestion outlining regenerative nodules (Fig. 5.8). Abundant lipofuscin is often found in the atrophic hepatocytes, and central veins may be distorted in a curvilinear fashion. Masson stain to confirm the relative lack of fibrosis and reticulin stain to highlight areas of two-cell thick plates indicative of hepatocyte regeneration (Fig. 5.9) are essential in establishing the diagnosis. While NRH is often reported in conjunction with portal vein changes of hepatoportal sclerosis, a minority of the CVID patients reported with NRH exhibit abnormal portal vein vasculature in the portal tracts (Malamut et al. 2008).
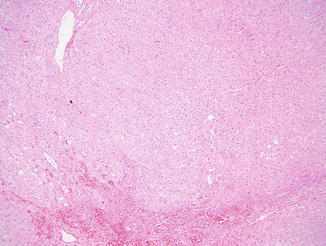
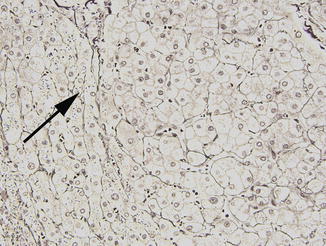
Fig. 5.8
Nodular regenerative hyperplasia is often inconspicuous on routine stains but may have a vaguely nodular appearance. Here sinusoidal congestion is seen in areas of hepatocyte atrophy
Fig. 5.9
Nodular regenerative hyperplasia. Reticulin stain is useful in showing alternating areas of thickened cell plates and hepatocyte atrophy (arrow)
An appreciable number of patients with CVID have a relatively mild chronic hepatitis pattern of injury on liver biopsy in the absence of hepatitis C infection (Daniels et al. 2009). Portal tracts contain a predominantly lymphocytic infiltrate, without mature plasma cells (Fig. 5.10), as would be expected in CVID. Many of the cases of mild predominantly lobular hepatitis were found to occur in patients with mucosal inflammation in GI biopsies, suggesting that the mild liver inflammation is a reaction to the inflammatory process in the GI tract (Daniels et al. 2009). Alternatively, the liver disease may be autoimmune in origin, as liver dysfunction in CVID patients is associated with peripheral blood lymphocyte abnormalities and other manifestations of autoimmunity (Malamut et al. 2008). Autoimmune hepatitis (AIH) in CVID is not distinguishable from other diseases causing a chronic hepatitis pattern of injury, and plasma cells are absent or found in very small numbers in the liver in AIH in these patients.
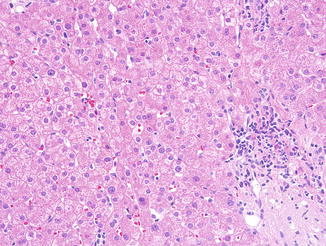
Fig. 5.10
Common variable immunodeficiency. A mild chronic hepatitis pattern of injury, with mononuclear portal inflammation with minimal interface hepatitis is relatively common in CVID
Hepatitis C infection in CVID patients generally produces chronic liver disease, with some patients proceeding to cirrhosis within 2–3 years after infection (Ermis et al. 2008; Gow et al. 2002). The histologic features are similar to those in immunocompetent patients (Fig. 5.11) (see Chap. 12) and are characterized by a lymphocytic portal inflammatory infiltrate with interface hepatitis and a variable degree of lobular activity in the form of acidophilic bodies and spotty hepatocyte necrosis. Sinusoidal lymphocytosis may be seen, and Kupffer cell aggregates are often found. As in AIH, plasma cells are not found in the inflammatory infiltrate in the portal tracts or in the lobule. Initial biopsies of CVID patients with chronic hepatitis C showed cirrhosis in roughly 40 % of patients in one series from patients infected in the 1980s (Bjoro et al. 1999).
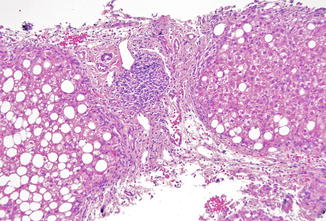
Fig. 5.11
Common variable immunodeficiency. Cirrhosis in CVID secondary to chronic hepatitis C shows features typical of the disorder in non-immunodeficient patients, with relatively mild activity in this example. Note nodular lymphoid aggregate and lack of plasma cells
Granulomas are found in the liver in 32–40 % of CVID patients who undergo liver biopsy (Ardeniz and Cunningham-Rundles 2009; Daniels et al. 2009). Many of these patients carry an additional diagnosis of sarcoidosis, and human herpes virus 8 has been detected by PCR and immunohistochemistry in lung and liver biopsies from patients with granulomatous or lymphocytic interstitial lung disease and granulomatous hepatitis (Wheat et al. 2005), suggesting a possible infectious etiology. The granulomas may be found in portal tracts or in the lobule and are typically non-necrotizing; mild portal fibrosis may be seen (Daniels et al. 2009), but the granulomas are typically not associated with fibrosis. In adults, primary biliary cirrhosis characterized by nonsuppurative, often granulomatous, destruction of small bile ducts may be seen, but this disorder essentially does not occur in the pediatric population, with only a small number of cases reported to date (Floreani et al. 2006; Dahlan et al. 2003).
5.1.1.2.4 Treatment
The mainstay of treatment for CVID is immunoglobulin replacement therapy, administered by an intravenous or subcutaneous route (Cunningham-Rundles 2010). However, IVIG is not effective in reducing GI symptoms due to its lack of IgA (Malamut et al. 2010; Oksenhendler et al. 2008). A prolonged course of metronidazole is needed for Giardia infection. For patients with a sprue-like lesion, gluten-free diet is usually ineffective. However, low-dose corticosteroids may induce significant reduction of diarrhea and malabsorption (Malamut et al. 2010). In severe cases, an element diet or total parental nutrition may be required. IVIG has not been shown to prevent or treat the IBD-like process in patients with CVID; instead, antibiotics, anti-inflammatory agents, or immunomodulators are used, as in the non-immunodeficient population (Yong et al. 2011). Infliximab has been shown to improve gut inflammation, but care should be taken to monitor for fungal infection in patients with severe T cell defects.
Because immunoglobulin products are pooled blood products derived from many donors, some patients have been exposed to hepatitis C through contaminated batches, most notably in 1994 (Rossi et al. 1997). The risk of viral hepatitis is considered to be much lower with third-generation IVIG products using viral inactivation and/or elimination steps such as low pH treatment, pasteurization, treatment with caprylic acid or solvent detergent, and nanofiltration methods (Radosevich and Burnouf 2010). However, given the risk of acquiring hepatitis, liver-related tests such as serum AST, ALT, and alkaline phosphatase levels are performed on a yearly or more frequent basis (Ward et al. 2008). Treatment for liver diseases arising in the setting of CVID is the same as in immunocompetent hosts. While hepatitis C infection may be more aggressive in the immunodeficient patient, sustained response to interferon and viral clearance have been reported in CVID (Bjoro et al. 1999; Castro et al. 1997), and liver transplantation may also be performed successfully (Cunningham-Rundles et al. 1999; Gow et al. 2002).
5.1.1.3 X-Linked Agammaglobulinemia
X-linked agammaglobulinemia (XLAG) is characterized by an inability to make antibodies to virtually all antigens, resulting in susceptibility to infections, encapsulated bacteria and enteroviruses such as polio virus. The typical XLAG patient is susceptible to bacterial infections due to absence of all circulating immunoglobulin subtypes; mature circulating B cells are low to absent.
The molecular basis of most cases of XLAG was elucidated in 1993, when a defect in the BTK (Bruton’s tyrosine kinase) gene was found (Tsukada et al. 1993; Vetrie et al. 1993). This gene encodes a non-receptor tyrosine kinase expressed in B cell and myelomonocytic cell lineages but not in T cells. Btk functions in intracellular signaling pathways essential for pre-B cell maturation, but the exact mechanism by which the defects in Btk lead to B cell maturation arrest remains unclear. XLAG may have more phenotypic diversity than has previously been recognized, as recently adults with mild or no clinical symptoms but with deficiencies in Btk have been described (Hashimoto et al. 1999; Mitsui et al. 2006; Valiaho et al. 2006).
5.1.1.3.1 Clinical Manifestations
GI manifestations are less common in XLAG than in CVID probably due to preservation of T cell functions (Agarwal and Mayer 2010; Marks et al. 2010). Age of onset of GI symptoms is younger than in CVID patients, and autoimmune diseases are less common. About one-third of patients present initially with GI complaints, most commonly diarrhea or perirectal abscess, and 10 % in one study had chronic GI symptoms, from persistent infection with Giardia lamblia, Salmonella species, or enteropathic E. coli or secondary to bacterial overgrowth. Chronic infection with rotavirus is also reported in this population (Saulsbury et al. 1980). However, no etiology for chronic diarrhea was found in half of these patients (Lederman and Winkelstein 1985). In addition to GI infections, patients with XLAG may develop a chronic ulcerating inflammatory condition clinically similar to Crohn’s disease, manifested by recurrent diarrhea, malabsorption, ulcers, and small bowel strictures (Cellier et al. 2000).
Patients with XLAG are at increased risk for malignancy, even in childhood. The most common malignancy in this group is non-Hodgkin’s lymphoma, often involving the GI tract; many of these cases occur in children under the age of 10 years (Lavilla et al. 1993). There are rare reported cases of gastric adenocarcinoma (Lavilla et al. 1993) and colorectal adenocarcinoma (van der Meer et al. 1993). The incidence of colorectal carcinomas for patients with XLAG is calculated as 30-fold, and the mortality as 59-fold greater than the normal European population (van der Meer et al. 1993). In most of the reported cases, XLAG patients with colorectal carcinoma are young adults in their 20s who present with advanced stage tumors. In one reported case, multiple colorectal adenomas in addition to carcinoma were found (van der Meer et al. 1993).
No specific hepatic diseases are characteristic of XLAG. Hepatitis C has been reported in 8 patients in a US registry of 201 patients (Winkelstein et al. 2006), with the source of infection traced to contaminated IVIG in 4 of these. While histologic features are similar to those of HCV in other populations, the severity of the necroinflammatory process is highly variable, ranging from severe (Fig. 5.12a, b) to minimal. A European study on surveillance of immunoglobulin safety in patients with immunodeficiency syndromes reports 11 XLAG patients positive for HCV RNA; 7 were asymptomatic, 3 had normal ALT levels, and 1 spontaneously recovered. Only 1 patient, who also had coexistent hepatitis B infection, died from hepatic failure, in contrast to CVID, in which 22 % of infected patients died from liver disease (Quinti et al. 2002). Fatal hepatitis from disseminated coxsackie virus and ECHOvirus (Bardelas et al. 1977) has also been reported in XLAG. In a case of hepatic veno-occlusive disease (VOD) leading to liver transplantation in an XLAG patient with human herpes virus 7 infection, examination of the explanted liver revealed intimal fibrosis and obliteration of terminal hepatic venules, with zone three hepatocyte necrosis and sinusoidal congestion (Soden et al. 2009).
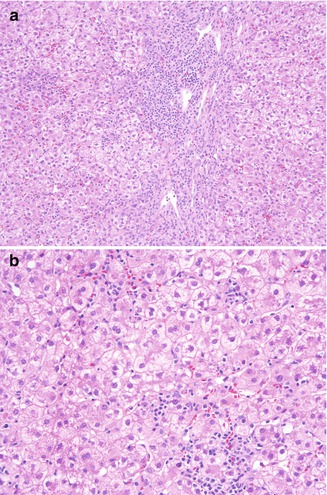
Fig. 5.12
X-linked agammaglobulinemia. Although chronic hepatitis C in XLAG may be milder than in CVID, in this example marked necroinflammatory activity with prominent interface hepatitis (a) and lobular activity (b) is present
5.1.1.3.2 Pathology
Small intestinal and colonic mucosal biopsies in the XLAG patient without GI symptoms are notable only for the lack of plasma cells in the lamina propria, giving the lamina propria an empty appearance (Fig. 5.13). No germinal centers are seen in gut-associated lymphoid tissues. Mucosal architecture is unremarkable, and villous blunting is not seen. Since biopsies are not routinely performed for chronic infection with rotavirus in patients with XLAG, descriptions of histopathologic findings are lacking, but moderate blunting of duodenal villi with crypt hyperplasia and an increase in lamina propria inflammatory cells are reported in acute infections (Davidson and Barnes 1979). Degenerative changes may be noted in epithelial cell on the surface of the villus, while crypt cells are spared and the crypt zone undergoes a compensatory hyperplasia. The histologic changes of acute rotavirus infection are reported to resemble celiac disease but are patchier and quickly revert to normal with resolution of infection (Moon 1997).
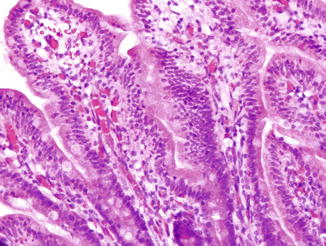
Fig. 5.13
X-linked agammaglobulinemia. No plasma cells are present in the lamina propria of the small intestinal mucosa
In biopsies from XLAG patients with chronic ulcerating GI inflammatory conditions, a prominent lymphocytic inflammatory infiltrate without plasma cells or granulomas is seen in the affected areas (Washington et al. 1996; Abramowsky and Sorensen 1988). In one case, enterovirus was found by PCR in inflamed ileum and adjacent mesenteric lymph nodes, suggesting that in some XLAG patients, infection may be responsible for these lesions (Cellier et al. 2000).
5.1.1.3.3 Treatment
The mainstay of treatment is gamma-globulin replacement to prevent bacterial infections. Adequate antibiotic administration with each infection is crucial for these patients.
5.1.1.4 X-Linked Hyper-IgM Syndrome
X-linked hyper-IgM syndrome (XHIM) is an uncommon primary immunodeficiency with X-linked inheritance, characterized by low serum concentration of IgG, IgE, and IgA and normal or elevated serum concentration of IgM due to defective immunoglobulin class switch recombination process (Davies and Thrasher 2010; Winkelstein et al. 2003). The annual incidence of XHIM is estimated as 1 in 517,000 live male births per year in the United States (Winkelstein et al. 2003).
XHIM is caused by mutations in the gene for CD40 ligand (Korthauer et al. 1993; DiSanto et al. 1993), leading to defects in B cell function. CD40 ligand is a small, 261-amino acid transmembrane protein expressed primarily on CD4+ T cells; it binds with CD40 on B cells, consequently promoting immunoglobulin isotype switching. T cells with mutated CD40 ligand gene do not interact with CD40 on the B cell surface, resulting in loss of isotype switching and consequently XHIM patients have very low levels of IgG and IgA and normal or elevated IgM levels. They are susceptible to pyogenic infections similar to those encountered in XLAG. Because CD40 is also expressed on macrophages/monocytes, CD40 ligand deficiency also affects T cell interaction with these cells, a process involving normal handling of opportunistic intracellular pathogens. Therefore, XHIM is also associated with an additional defect of cell-mediated immunity and a consequent susceptibility to opportunistic infections (Davies and Thrasher 2010). A variety of intracellular pathogens such as various mycobacterial species, fungi, and viruses (cytomegalovirus, adenovirus) are implicated in causing disease in these patients. The disorder is manifested clinically by recurrent bacterial infections typical of hypogammaglobulinemia, most commonly of the upper and lower respiratory tract, and neutropenia and lymphadenopathy (Levy et al. 1997). Infections with a wide variety of bacteria, fungi, viruses, and parasites reflect defects in T cell function as well as the resulting hypogammaglobulinemia.
5.1.1.4.1 Clinical Presentation
XHIM presents early in life, with over half of patients symptomatic by 1 year and almost all by age 4 (Winkelstein et al. 2003). The most common site of bacterial infection is the upper (~88 %) and lower respiratory tract (~83 %) (Levy et al. 1997).. Opportunistic infections with Pneumocystis jiroveci and Cryptosporidium parvum are common (Winkelstein et al. 2003; Levy et al. 1997; Wolska-Kusnierz et al. 2007). Disseminated infection (Hostoffer et al. 1994) and esophageal infection (Tu et al. 1991) with Histoplasma capsulatum have been reported. Patients with this disorder also develop autoimmune inflammatory disorders and cancer.
Diarrhea occurs in over half of patients and follows a chronic course in most (Levy et al. 1997). Chronic watery diarrhea due to Cryptosporidium infection occurs in approximately 25 % patients; Giardia intestinalis, Salmonella, and Entamoeba histolytica have also been implicated. However, in more than 50 % of patients with recurrent or protract diarrhea, no causative agents can be detected. Nodular lymphoid hyperplasia involving the GI tract is reported in ~5 % of patients (Levy et al. 1997). Lymphoid hyperplasia may also result in hepatosplenomegaly, lymphadenopathy, and enlargement of the tonsils (Ramesh et al. 1998). Patients with this disorder are prone to autoimmune hematologic diseases, including cyclical or chronic neutropenia, and oral and perianal ulcers are common during neutropenic episode. Inflammatory bowel disease has been reported in two patients with XHIM and chronic diarrhea; clinical details are not provided (Levy et al. 1997).
Malignancies of the liver and GI tract including hepatocellular carcinoma, biliary tract cancer, and poorly differentiated neuroendocrine carcinoma (usually small cell carcinoma) of the gastroenteropancreatic system (Nagaraj et al. 2007; Malhotra and Li 2008; Zirkin et al. 1996) commonly occur in adolescents and young adults with XHIM and account for approximately 25 % of the mortality associated with the disease.
5.1.1.4.2 GI Pathology
Patients with XHIM are particularly susceptible to Cryptosporidium infection, which is frequently associated with chronic diarrhea and sclerosing cholangitis (Davies and Thrasher 2010; Winkelstein et al. 2003). The parasite can infect epithelial cells of the entire GI tract and bile duct. Cryptosporidium infection is usually diagnosed by examination of stool and bile samples by modified Ziehl-Neelsen technique, by immunofluorescence assay, or by molecular analysis followed by genotyping. GI biopsy is rarely needed for the diagnosis. In CD40 ligand knockout mice, the occurrence of C. parvum in the gall bladder was consistently associated with the presence of a mononuclear infiltrate in the underlying tissue. However, there was no inflammatory infiltrate in the lamina propria or muscularis propria of the infected bowel wall (Cosyns et al. 1998). Similarly, in patients with GI cryptosporidiosis, 2- to 5-micron basophilic spherical structures are seen attached to microvillus surface of epithelium; significant inflammation is always absent (Fig. 5.14). However, variable villous abnormality or eosinophilic infiltrates in the intestinal mucosa may be seen. Massive proliferation of IgM-producing plasma cells may involve the GI tract, liver, and gallbladder, usually in the second decade of life, and may prove fatal (Rosen et al. 1995).
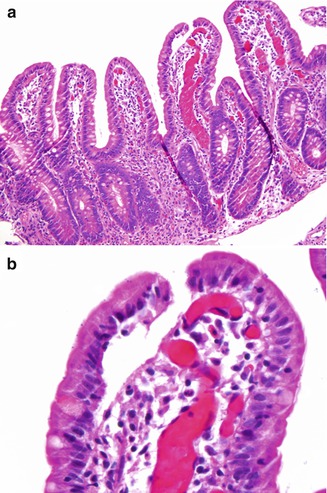
Fig. 5.14
X-Linked hyper-IgM syndrome. Mild villous blunting and increased lamina propria eosinophils are present in this duodenal biopsy (a). Cryptosporidium oocytes are seen attached to microvillus surface of epithelium (b)
5.1.1.4.3 Liver Pathology
Data from European and US registries of XHIGM indicate that up to half of XHIGM patients have abnormalities in liver tests. Liver disease in one study of 53 patients showed age-related progression and was not seen in patients who died before age 5 (Hayward et al. 1997). However, of the 16 patients in this study who reached the age of 20, only 4 did not have liver disease. Biliary tract infection with Cryptosporidium leading to cholangitis or cirrhosis is relatively common in XHIGM patients. Sclerosing cholangitis, with degenerative changes in bile duct epithelium and periductal edema and fibrosis, is seen on liver biopsy (Fig. 5.15) and is associated with Cryptosporidium infection in over half of the reported cases (Davies and Thrasher 2010; Winkelstein et al. 2003; Levy et al. 1997; Hayward et al. 1997).
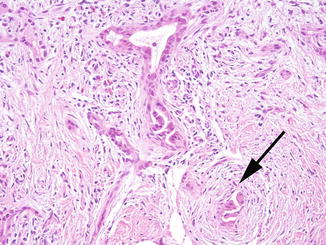
Fig. 5.15
Severe combined immunodeficiency. Sclerosing cholangitis in SCID and other immune deficiencies is characterized by bile duct distortion and loss, and in this example, periductal fibrosis (arrow). This pattern of injury is often associated with biliary cryptosporidial infection
Viral hepatitis leading to cirrhosis is also reported in the XHIGM population. Nine of 56 European patients in one study had chronic hepatitis, 5 with hepatitis B, and 1 with hepatitis C. The two patients in this series with HBV infection both died of hepatocellular carcinoma (Levy et al. 1997). A similar number of cases with hepatitis are reported from the US registry (Winkelstein et al. 2003); unusual causes of hepatitis in this series include ECHOvirus, histoplasmosis, and Bartonella.
In addition to hepatocellular carcinoma arising in the setting of chronic liver disease, cholangiocarcinomas and gallbladder cancers associated with biliary tract Cryptosporidial infection are reported. At least eight high-grade neuroendocrine carcinomas involving the liver or pancreas have been reported in adolescents and young adults with XHIGM (Erdos et al. 2008; Seyama et al. 1998). In some cases, the tumors are so bulky and widespread at the time of diagnosis that the primary site cannot be ascertained (Erdos et al. 2008). These tumors grow primarily in solid nests and sheets but may show rosette formation (Hayward et al. 1997) and are positive for synaptophysin and CD56. Expression of chromogranin A is variable. Outcome of these malignancies is generally poor in this population, with most patients succumbing rapidly.
5.1.1.4.4 Treatment
Immunoglobulin replacement therapy is the mainstay of treatment for XHIM (Davies and Thrasher 2010). A number of XHIM patients have been treated with stem cell transplantation. Appropriate antimicrobial agents are used for infections, and immunosuppressants for autoimmune disorders (Davies and Thrasher 2010). Antibiotic prophylaxis, IVIG, and routine childhood immunization with killed vaccines are used to prevent infections. The availability of nitazoxanide may help treat Cryptosporidium infection and prevent biliary complications (Winkelstein et al. 2003).
5.1.1.5 Hyperimmunoglobulin E Syndrome (Job’s Syndrome)
Hyperimmunoglobulin E syndrome (HIES) is a rare primary immunodeficiency characterized by susceptibility to staphylococcal infectious, atopic dermatitis, and extremely high serum IgE levels. Serum concentrations of other immunoglobulins (IgG, IgA, IgM, and IgD) and IgG subclasses are normal (Minegishi 2009). Two inheritance patterns have been described: (1) autosomal dominant (classic) HIES with abnormalities of bone and connective tissue (Minegishi et al. 2007) and (2) autosomal recessive (novel) form of HEIS associated with susceptibility to intracellular bacterial and viral infections (Minegishi et al. 2006). However, most of cases are sporadic and present with the classic multisystem HIES.
Recently, a dominant-negative mutation in the signal transducer and activator of transcription 3 (STAT3) gene has been identified in patients with sporadic HIES (Minegishi et al. 2007). STAT3 is a transcript factor that is activated in responses to many cytokines and growth factors. Activated STAT3 binds to the STAT3-response elements in promoters of various genes including those regulating cytokine-mediated generation of TH17 cells, a newly identified subset of helper T cells critical in the clearance of fungal and extracellular bacterial infection. A TH17 defect in some subjects with autosomal dominant HIES (Milner et al. 2008) has also been identified. Mutations in the STAT3 gene in HIES patients occur in the DNA-binding domain, which diminishes its DNA-binding ability, and cause defective response to many cytokines, consequently resulting in impaired TH17 cell differentiation. The inability to produce TH17 cells due to STAT3 mutation is one of the primary mechanisms underlying susceptibility to the recurrent infections commonly seen in HIES. In contrast, a tyrosine kinase 2 (TYK2) deficiency and defective DOCK8 were identified as underlying mechanisms for autosomal recessive HIES (Minegishi et al. 2006). TYK2 transduces a signal downstream from a number of cytokines including IL-10, IL23, IL6, IL-12, and IFNα. TYK2 mutations lead to a defect in signal transduction from IL12 and IFNα, thus resulting in susceptibility to intracellular bacterial and viral infections. In addition, defective IL6 and IL23 result in impaired TH17 function, thereby leading to susceptibility to fungal and extracellular bacterial infection (Minegishi 2009).
5.1.1.5.1 Clinical Presentation
Patients with HIES present with recurrent staphylococcal skin abscesses, recurrent pneumonia with pneumatocele, elevated serum IgE, eczema, candidiasis, and eosinophilia (Grimbacher et al. 1999). In addition to findings related to the immune system, characteristic facial features and dental and skeletal abnormalities have been reported in patients with classical HIES (Grimbacher et al. 1999). Patients with the novel variant of HIES have no skeletal abnormalities but suffer from recurrent viral infections (Minegishi et al. 2006). Chronic diarrhea and disorders resembling inflammatory bowel disease are not reported in HIES, and GI manifestations of the disease appear limited to opportunistic infections. Perforation of the colon, probably related to infection with staphylococcal species, has also been reported (Hwang et al. 1998). Patients with HIES do not appear to be at increased risk for primary GI malignancy; however, associations with lymphomas of both B and T cell derivation have been reported (Leonard et al. 2004; Onal et al. 2006). HIES is not associated with specific types of liver injury.
5.1.1.5.2 Pathology
Mucocutaneous candidiasis and tissue-invasive fungal infections with Cryptococcus have been reported in the esophagus (Jacobs et al. 1984) and colon (Hutto et al. 1988). Mycobacterial infections are uncommon but have been reported. Patients with mycobacterial infection can present with ulcerative lesions affecting the large bowel and causing GI bleeding. Histological examination shows accumulation of macrophages containing acid fast bacilli (Gotzberger et al. 2004). Ileocecal histoplasmosis mimicking Crohn’s disease (Alberti-Flor and Granda 1986; Steiner et al. 2009) has also been described in patients with HIES. Colonoscopy showed edema, erythema, and friability of the mucosa of the cecum and ileocecal valve and linear ulcerations in the terminal ileum. Histological examination revealed chronic inflammation, prominent granuloma formation, and macrophages containing budding yeasts.
5.1.1.5.3 Treatment
High-dose IVIG is effective in the treatment of severe eczema in patients with HIES and atopic dermatitis (Freeman and Holland 2008). Chronic antibiotics are used to protect HIES patients from staphylococcal infection. Patients with other infections are treated with standard antibiotic regimens.
5.1.2 Combined Cellular/Humoral Immunodeficiencies
5.1.2.1 Severe Combined Immunodeficiency
Severe combined immunodeficiency (SCID) is a heterogeneous group of congenital disorders characterized by defects in both B and T cell function and is the most severe form of primary immunodeficiency. SCID patients present with severe, recurrent infections in early infancy after they lose protection by maternal antibodies (Stephan et al. 1993).
A number of molecular defects may result in SCID. Most are autosomal recessive; these include adenosine deaminase (ADA) deficiency, accounting for 50 % of autosomal recessive SCID, purine nucleoside phosphorylase (PNP) deficiency, T cell receptor deficiencies, Zap70 deficiency, JAK3 deficiency, and IL-7 receptor deficiency (Jones and Gaspar 2000). ADA and PNP are involved in nucleotide metabolism and salvage. Their deficiencies lead to accumulation of toxic metabolites, to which lymphocytes are exquisitely sensitive. Both T and B lymphocytes are affected. X-linked SCID resulting from a defect in the common γ chain caused by mutations in the IL2RG gene is the single most common type of SCID (Noguchi et al. 1993). This type of SCID has a characteristic phenotype of absence of T and NK cells but normal B cell numbers, though these are dysfunctional (Jones and Gaspar 2000). In addition, a subset of patients with SCID have mutations in genes involved in V(D)J recombination, including the recombination activating genes (RAG1 and RAG2), Artemis, DNA protein kinase, and LIG4 (van der Burg and Gennery 2011). V(D)J recombination takes place in developing B and T cells and is responsible for the rearrangement of the immunoglobulin and T cell receptor genes. Therefore, SCID patients with mutations in one of these genes have a phenotype of absence of both T and B lymphocytes.
5.1.2.1.1 Clinical Presentation
Children with SCID typically present within the first few months of life with persistent respiratory tract and GI infections (Stephan et al. 1993; Fischer 2000). Affected children usually suffer from persistent viral diarrhea and failure to thrive. A variety of other infectious pathogens, including Salmonella, Escherichia Coli, or Cryptosporidium, may also cause chronic GI infection in SCID patients. Oral, esophageal, and perianal candidiasis is common (Stephan et al. 1993). Children with SCID may be at a greater risk for reflux esophagitis than the normal population (Boeck et al. 1997). SCID patients receiving nonirradiated blood products or those receiving allogeneic bone marrow transplants are susceptible to graft-versus-host disease (GVHD) (Stephan et al. 1993; Snover et al. 1985). Patients with SCID may be at increased risk for primary GI malignancy, especially B cell lymphoma.
5.1.2.1.2 Pathology
In general, GI biopsies from these patients show a hypocellular lamina propria without plasma cells or lymphocytes (Fig. 5.16a). Because these patients are susceptible to viral infections, examination of stool for viral particles may be indicated. In particular, rotavirus, normally a self-limited infection, may cause chronic diarrhea in these children. Although villous blunting has been described in acute rotavirus infection in normal children (Moon 1997) and in animal models (Salim et al. 1995), the intestinal pathology of chronic rotavirus infection in SCID patients has not been described. Cytopathic viral infections that may be identified on GI biopsy include cytomegalovirus and adenovirus (Fig. 5.16b). Furthermore, a GVHD-like process affecting the colon and small intestine has been described in patients with SCID who had not undergone bone marrow transplantation (Snover et al. 1985; Lee et al. 1991).
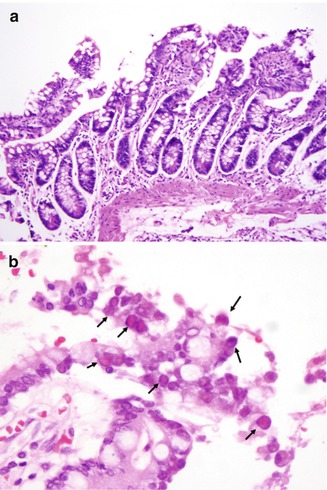
Fig. 5.16
Severe combined immunodeficiency. Small intestinal biopsy is featured by a hypocellular lamina propria (a). Intestinal biopsy from an SCID patient shows adenoviral nuclear inclusions (b) (arrows)
5.1.2.1.3 Liver Disease in Severe Combined Immunodeficiency
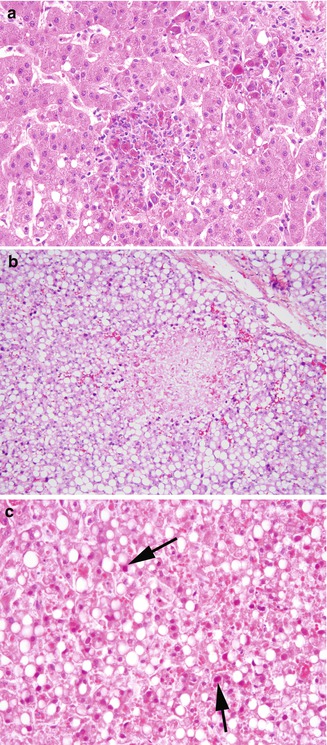
Most liver disease reported in severe combined immunodeficiency in older studies was iatrogenic in nature, with graft-versus-host disease from stem cell transplantation or nonirradiated blood products, and cholestatic changes due to total parenteral nutrition common findings in biopsies, and veno-occlusive disease related to marrow ablative regimens in autopsies (Washington et al. 1993). Fatal adenovirus hepatitis as part of systemic adenoviral infection also occurs in this population (Washington et al. 1993), and cytomegalovirus is also reported (Fiore et al. 1998). In adenoviral hepatitis, the liver shows patchy (Fig. 5.17a, b) or confluent areas of hepatocyte necrosis, with little inflammatory response. The nuclei of degenerating hepatocytes at the periphery of necrotic areas are characteristically hyperchromatic and homogeneous due to accumulation of the virus; these cells are known as “smudge cells” (Fig. 5.17c).
 < div class='tao-gold-member'>
< div class='tao-gold-member'>




Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
