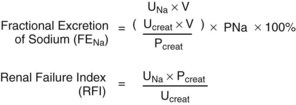
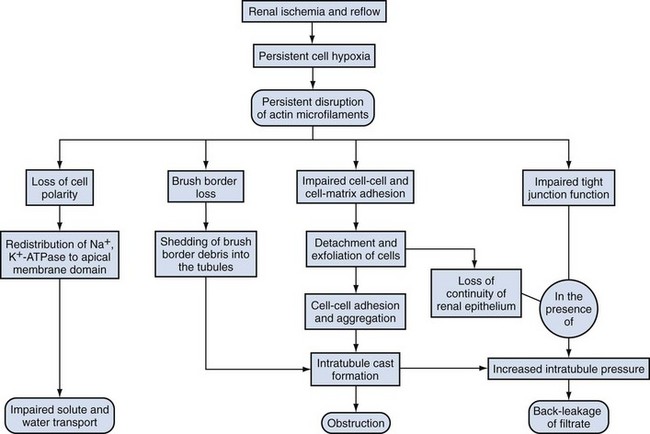
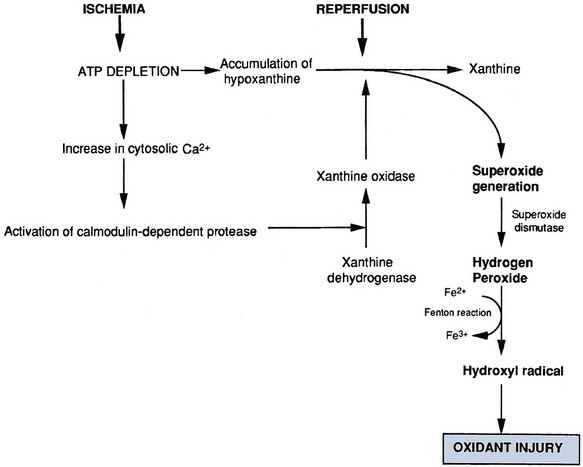
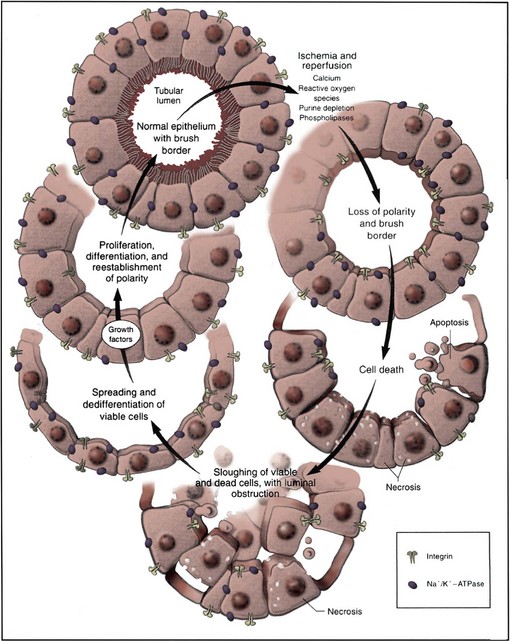
David A. Goldfarb, MD, Emilio D. Poggio, MD Disorders of renal function are ubiquitous in the contemporary practice of medicine. Current estimates suggest that kidney disease affects approximately 10% to 13% of the adult American population (Coresh et al, 2007), and thus awareness of the diagnosis and management of this disease has been the focus of the National Kidney Foundation (NKF) and other health care organizations (www.kidney.org). In 2002 the NKF published the Kidney Disease Outcomes Quality Initiatives (K/DOQI) Guidelines setting up the framework for the definition, diagnosis, classification, and management of kidney disease. These guidelines broadly grouped kidney disease into two major categories: acute kidney injury (AKI) and chronic kidney disease (CKD). These conditions are common in clinical practice, so urologists are likely to encounter issues related to renal function and its aberration on a daily basis. This chapter is designed to give a contemporary insight into the nature of kidney disease and its treatment. Acute kidney injury (AKI) is a common problem in the contemporary practice of medicine and urology. Prospective studies have demonstrated that 2% to 5% of all patients admitted to a general medical/surgical hospital unit will develop AKI (Nolan and Anderson, 1998). In selected patients in the intensive care unit following cardiovascular or abdominal vascular surgery, the incidence may exceed 20%. Epidemiologic studies continue to indicate that despite advances in the overall medical care of patients, the development of AKI is associated with significant increases in morbidity (which prolongs hospitalization and increases costs) and mortality (Dimick et al, 2003). The high occurrence and substantial morbidity and mortality of AKI demand a logical approach to its early recognition and prevention, as well as prompt diagnosis and management of its complications. The cardinal feature of AKI is a decline in glomerular filtration rate (GFR). Although ideally determined by inulin or radio-isotopic clearance techniques, it is usually identified in routine clinical practice by a rise in serum blood urea nitrogen (BUN) or creatinine. It is important to understand the limitations of these common clinical chemistries in order for them to be properly interpreted. The correlation among BUN, creatinine, and GFR assumes they are delivered into the serum at a constant rate. Therefore conditions such as hypercatabolic state and massive trauma, which may be seen in surgical patients, can affect renal function assessment. BUN may be disproportionately elevated in states of marked volume contraction, hypercatabolic states, and with marked increases in protein loads seen with gastrointestinal bleeding or total parenteral nutrition (TPN). SCr is produced at a constant rate by muscle and more accurately reflects GFR. When renal function deteriorates, tubular secretion represents an increasing proportion of creatinine excretion. Therefore creatinine clearance may overestimate GFR as renal function slowly declines when measured in the “steady state.” Both the creatinine clearance and the Cockcroft Gault calculation may be useful tools to estimate GFR because they take into account the patient’s body size, age, and SCr. For example, the Cockcroft Gault formula states that creatinine clearance = (140 − age) × weight × (0.85, female) divided by 72 × plasma creatinine (Cockcroft and Gault, 1976). Both methodologies have their disadvantages. The urinary creatinine clearance method is cumbersome, and the Cockcroft Gault method may have a margin of error up to approximately 30% compared with GFR measured by formal I125 iothalamate techniques. Recently the Modification of Diet in Renal Disease (MDRD) calculations have been used as a way to estimate GFR. This equation has then been reformulated to account for various creatinine assay calibration biases across different laboratories (Levey et al, 2007). However, none of these techniques are suitable for estimating GFR in the AKI patient because such a “steady state” does not exist (Hoste et al, 2005). Use of “real-time” estimates of GFR using infused radio-isotopic plasma disappearance techniques in the ICU patient with AKI has been advocated by some but has not achieved wide clinical acceptance. In clinical practice the experienced clinician infers from a rapidly rising SCr and clinical picture that the patient’s true GFR is approaching zero. Clinically, it is useful to separate the causes of AKI into three major categories: prerenal, intrarenal, and postrenal. Distinguishing among the three basic categories of AKI is a challenging clinical exercise. Assigning a patient to one of the three categories usually requires a combination of clinical and laboratory evaluations and may require invasive monitoring of central hemodynamics or imaging studies of the genitourinary tract. The importance of differentiating the major causes of AKI must be stressed because the initial evaluation and management are tailored to the particular cause. Because the greatest proportion of hospital-acquired AKI is secondary to acute tubular necrosis (ATN), this chapter places special emphasis on the diagnosis, pathophysiology, and management of ATN. For example, a report from Madrid evaluated 748 cases of AKI at 13 tertiary care hospital centers (Di Tullio et al, 1996). The causes of AKI were: 45% ATN, 21% prerenal, 13% acute or chronic renal failure, 10% urinary tract obstruction, 4% glomerulonephritis/vasculitis, 2% acute interstitial nephritis (AIN), and 1% atheroembolic renal disease. Earlier reports had documented that AKI occurs more commonly in the surgical/trauma ICU, medical ICU, and postoperative units. A recent study of the American College of Surgeons National Surgical Quality Improvement Program has identified 11 independent preoperative predictors for the development of AKI including age 56 years or older, male sex, emergency surgery, intraperitoneal surgery, diabetes mellitus necessitating oral therapy, diabetes mellitus necessitating insulin therapy, active congestive heart failure, ascites, hypertension, mild preoperative renal insufficiency, and moderate preoperative renal insufficiency (Kheterpal et al, 2009). Under normal circumstances, the kidney can maintain normal renal blood flow (RBF) and GFR down to perfusion pressures of approximately 60 mm Hg. The phenomena of autoregulation require a complex interaction of physiologic factors to maintain RBF and GFR. In some hospitalized patients with disordered autoregulation, a reduction in RBF and GFR may occur with modest or even no discernable fall in systemic blood pressure (BP). In the setting of decreased renal perfusion, angiotensin II (AII) and vasodilatory prostaglandins play an important role in maintaining glomerular hydrostatic pressure and GFR. The three major determinants of GFR are renal plasma flow, glomerular hydrostatic pressure, and glomerular permeability. AII has selectively greater vasoconstrictor effects on the efferent than afferent arteriole, whereas vasodilatory prostaglandins can cause afferent arteriolar vasodilation. Drugs that selectively block AII synthesis, angiotensin-converting enzyme inhibitors (ACE inhibitors), and AII receptor blockers (angiotensin II receptor blockers or ARBs) or that inhibit vasodilatory prostaglandin synthesis such as nonsteroidal anti-inflammatory drugs (NSAIDs) (Whelton, 1999) may cause AKI. This is prone to occur in clinical settings when GFR is already compromised (Toto et al, 1991; Whelton, 1999). Prerenal azotemia may be encountered in both the volume-depleted and volume-overloaded patient (Table 43–1). True volume depletion may result from renal or extrarenal losses that result in systemic hypotension and renal hypoperfusion. In the volume-overloaded patient, with edematous states such as cirrhosis and congestive heart failure, prerenal azotemia may occur because the kidney perceives that the vascular tree is underfilled (i.e., “ineffective arterial blood volume”). This results in renal hypoperfusion. Prerenal azotemia may also occur owing to high-grade bilateral renal artery stenosis or in states of renal hypoperfusion due to redistribution of extracellular fluid with peripheral vasodilation as seen with sepsis. As noted earlier, ACE inhibitors/angiotensin II receptor blockers (ARBs) and NSAIDs may alter the vasoconstrictor effects of AII and the vasodilatory effects of prostaglandins to produce prerenal azotemia. This is especially the case in elderly patients with impaired GFR, subtle volume depletion, or occult renal artery stenosis. Table 43–1 Prerenal Causes of Acute Kidney Injury The hepatorenal syndrome (HRS) represents a unique, severe form of prerenal azotemia. HRS refers to the development of AKI in the patient with advanced hepatic disease, often due to cirrhosis but also seen with metastatic tumor or alcoholic hepatitis. The reduction in renal perfusion appears to relate to relative splanchnic vasodilatation, which may be mediated via nitric oxide, the endothelium-derived relaxing factor (Martin et al, 1998; Gines and Arroyo, 1999). HRS is characterized by oliguria, a benign urinalysis, urinary sodium avidity, and a progressive rise in SCr (Cardenas, 2005). HRS is a prerenal disease because the kidneys are normal histologically and have been successfully used in renal transplantation (Koppel et al, 1969). The diagnosis is one of exclusion, after ATN, acute glomerulonephritis, vasculitis, or correctable forms of reduced renal perfusion have been excluded. Thus the diagnosis of HRS requires a lack of improvement in renal function following discontinuation of potential nephrotoxins and a trial of fluid repletion. The best hope for reversal of HRS is improvement in hepatic function or successful liver transplantation (Gonwa et al, 1991; Cardenas, 2005); however, not all cases of HRS will resolve with this approach (Pham et al, 2005). The utilization of simultaneous liver-kidney transplantation is evolving. It is difficult to predict which cases of HRS will regain renal function spontaneously with liver transplant alone versus those that have irreversible renal injury and would benefit from a kidney transplant. Protracted HRS may lead to irreversible loss of kidney function. There has recently been a consensus conference that has defined criteria to proceed with kidney transplantation in the setting of HRS (Eason et al, 2008). Results of medical interventions have been disappointing. Therapy with ACE inhibitors has been complicated by systemic hypotension and reductions in GFR (Gentilini et al, 1993). The ADH analog ornipressin, which should decrease splanchnic dilatation, may also induce renal ischemia (Guevara et al, 1998; Gulberg et al, 1999). Treatment with a prostaglandin analog, misoprostol, has yielded conflicting results, but preliminary evidence with N-acetylcysteine suggested improvement in splanchnic dilatation and nitric oxide production in a small series of HRS patients (Holt et al, 1999). Similarly, combination therapy with midodrine (a selective α1-adrenergic agonist) and octreotide (a somatostatin analogue) has yielded preliminary information that it may be safe and effective as compared with renal-dose dopamine in a small series of HRS patients (Angeli et al, 1999). Terlipressin, a vasopressin analog available in Europe, is also showing promising results in small studies that need to be confirmed in larger multicenter trials (Fabrizi et al, 2006; Alessandria et al, 2007; Martin-Llahi et al, 2008; Sanyal et al, 2008; Karwa and Woodis, 2009; Skagen et al, 2009). Nevertheless, overall the impact of medical therapy for HRS has been minimal and the outlook for HRS without orthotopic liver transplantation appears bleak. The role of hemodialysis support appears to be limited to those HRS patients awaiting liver transplantation or resolution of their primary hepatic disease because survival is generally limited by the severity of liver failure. Hemodialysis is often difficult in HRS because of hemodynamic instability. Obstruction of the urinary tract may cause AKI. To be the cause of AKI, urinary tract obstruction must involve the outflow tract of both kidneys, unless preexisting renal dysfunction is present, in which case the obstruction may involve only a single kidney. Patients with acute urinary tract obstruction may present with hematuria, flank or abdominal pain, or signs of uremia. A high index of suspicion for urinary tract obstruction should exist for patients with prior abdominal or pelvic surgery, neoplasia, or radiation therapy. Although oligoanuria suggests complete obstruction, partial obstruction may exist in the presence of adequate urinary output. Oligoanuria is a powerful diagnostic clue that suggests a differential diagnosis of urinary tract obstruction, severe ATN with cortical necrosis, or bilateral vascular occlusion. Lesions that may cause obstruction can be either intrinsic or extrinsic to the genitourinary tract. If urinary tract obstruction is a diagnostic consideration, the renal ultrasound is sensitive and specific (90% to 95%) in confirming the diagnosis of hydronephrosis. This test may be operator dependent, so the experience of the radiologist is crucial. False-negative tests may be seen with periureteral metastatic disease or retroperitoneal fibrosis (Somerville et al, 1992). Renal radionuclide studies or retrograde pyelography may be helpful in this circumstance. If urinary tract obstruction is a diagnostic consideration, renal ultrasound should be performed because obstruction represents a potentially reversible cause of AKI. The combination of AGN (based on urinalysis findings) and a rapid loss of kidney function defines the clinical syndrome of rapidly progressive glomerulonephritis (RPGN). The differential diagnosis and management of RPGN are beyond the scope of this chapter (Lee et al, 2004). A simplified differential diagnosis of RPGN is summarized in Table 43–2. It is crucial to appreciate the impact that urinary microscopic findings of AGN have on the aggressive evaluation and management of patient with this type of AKI. This usually includes a renal biopsy and detailed serologic evaluation for the presence of systemic vasculitis, collagen vascular disease, and an infectious process. RPGN comprises a group of glomerulonephritides that progress to renal failure in a matter of days to months in the presence of extensive extracapillary proliferation (i.e., crescent formation in a large percent of glomeruli). Patients with RPGN have been divided into three patterns defined by their immunologic pathogenesis: (1) type I: antiglomerular basement membrane (anti-GBM) (e.g., Goodpasture); (2) type II: immune complex deposition disease (e.g., systemic lupus erythematosus, poststreptococcal glomerulonephritis); and (3) type III: pauci-immune (e.g., antineutrophil cytoplasmic autoantibody [ANCA]–positive disease such as Wegener granulomatosis). Patients are categorized on the basis of the results of the immunofluorescence of the renal biopsies and results of serologic testing for anti-GBM titer, ANCA, lupus serologies, and so on. Specific therapies (including parenteral steroids, cyclophosphamide or other cytotoxics, and possibly plasma exchange) tailored to the disease entity diagnosed may be lifesaving. Hence early recognition of RPGN based on urinalysis findings is critical. Table 43–2 Differential Diagnosis of Rapidly Progressive Glomerulonephritis The diagnosis of AKI secondary to AIN may be suggested by the urinalysis findings of sterile pyuria, white blood cell casts, and eosinophiluria (using Hansel stain) (Michel and Kelly, 1998). AIN is most often induced by drug therapy, although sarcoidosis, streptococcal, viral, or Legionella infections may also be responsible. The list of offending drugs associated with AIN is extensive, but the most common causes of AIN include those identified in Table 43–3. The major histologic changes are interstitial edema and marked interstitial infiltrate of T lymphocytes and monocytes (Laberke and Bohle, 1980). Eosinophilic plasma cells and polymorphonuclear cells may also be detected. Granulomata formation, once thought particular to the renal disease of sarcoidosis, can occur in any form of AIN. Table 43–3 Drugs That Most Commonly Cause Acute Interstitial Nephritis The clinical presentation, although variable, usually involves an abnormal urine sediment (described earlier), fever, and a rising SCr associated with the administration of the offending drug (Nolan et al, 1986). Skin rash is seen in about 25% of cases. Eosinophilia and eosinophiluria is present in more than 75% of cases, with the exception of AIN due to NSAIDs, where fever, rash, and eosinophilia are typically absent. Proteinuria with most drugs is usually modest with less than 0.5 to 1 g/day. Proteinuria in the nephrotic range has been frequently seen with AIN of NSAIDs (especially fenoprofen) and in selected cases with ampicillin, rifampin, ranitidine, and interferon. It is speculated that increased glomerular capillary permeability is related to cytokine release of the infiltrating T cells (Neilson, 1989). The development of AIN is not dose dependent, and recurrence can occur with second exposures to the same or related drug. The onset of AIN may occur from 3 to 5 days (especially second exposures) to several weeks after drug therapy. The diagnosis is usually suspected in the AKI patient with characteristic urinary sediment abnormalities and a history of an offending drug therapy. Although the clinical picture may be highly suggestive of AIN, the diagnosis is confirmed only by renal biopsy. Most clinicians will observe the response to withdrawing the offending agent (Kida et al, 1984). No further evaluation or therapy is required if renal function begins to improve in several days. Lack of response, severe AKI, or uncertainty of diagnosis may be indications for definitive diagnosis with renal biopsy. Initial therapy consists of discontinuing the offending drug with the expectation that renal function will begin to improve in 3 to 7 days (Baker and Pusey, 2004). There are no controlled trials evaluating the efficacy of immunosuppressive therapy in the small number of patients reported with biopsy-proven AIN who did not respond to expectant management. There is some experimental and suggestive clinical evidence that steroid and/or cytotoxic therapy may be beneficial to hasten recovery of renal function and reduce interstitial fibrosis. Overall, AKI may affect 2% to 7% of patients in a tertiary care hospital, and the incidence of AKI in the surgical or medical ICU may exceed 25% to 35% (Palevsky et al, 2008). The majority of all hospital-acquired AKI is secondary to ATN (Myers and Moran, 1986; Uchino et al, 2005). Renal hypoperfusion and renal ischemia are the most common causes of ATN, although nephrotoxic insults from various agents are being recognized with increasing frequency. A detailed listing of both exogenous and endogenous nephrotoxic compounds is summarized in Tables 43–4 and 43–5. Table 43–4 Causes of Exogenous Toxic Acute Kidney Injury * Direct toxicity or indirect systemic effects (shock, intravascular hemolysis, or coagulation). † Slow onset of renal failure unless associated with rhabdomyolysis. From Nally JV. Acute renal failure. In: Stoller JK, Ahmed M, Longworth DL, editors. The Cleveland Clinic intensive review of internal medicine. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2000. p. 568. Table 43–5 Acute Kidney Injury Related to Endogenous Nephrotoxic Products * Questionable direct nephrotoxic effect. From Nally JV. Acute renal failure. In Stoller JK, Ahmed M, Longworth DL, editors. The Cleveland Clinic intensive review of internal medicine. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2000. p. 567. Pigment nephropathy may be suspected in the appropriate clinical situation (post-traumatic or atraumatic after intoxications) where there is discrepancy between the finding of hematuria by dipstick and the absence of red blood cells on urinary microscopy. In urology, two clinical circumstances have been identified in association with rhabdomyolysis. This includes protracted extended lithotomy positioning (as seen in urethral stricture cases) (Anema et al, 2000) and after laparoscopic donor nephrectomy (Kuang et al, 2002; Troppmann and Perez, 2003; Reisiger et al, 2005). The combination of renal hypoperfusion and the nephrotoxic insult of myoglobin or hemoglobin within the proximal tubule can result in ATN. Early recognition of this disorder is crucial because a forced alkaline diuresis is indicated to minimize nephrotoxicity. Similarly, the tumor lysis syndrome may be suspected in the appropriate clinical setting, when marked hyperuricemia/hyperuricosuria and crystalluria are recognized. A forced alkaline diuresis may limit nephrotoxicity and is usually recommended prophylactically before an aggressive chemotherapy regimen. The list of potential exogenous nephrotoxic agents is exhaustive (see Table 43–4). Simply stated, a patient who develops ATN while receiving medications should have each medication reviewed for the possibility of nephrotoxicity. The most commonly seen nephrotoxins in the hospitalized patient include radiographic contrast material, antibiotics (especially aminoglycosides and amphotericin B), chemotherapeutic agents, NSAIDs, and ACE-inhibitor drugs. In the contemporary practice of hospital-based medicine, recognition of AKI in HIV patients deserves special comments. Patients with HIV-infection may develop AKI due to the same causes as uninfected patients (Cohen et al, 2008), but protease inhibitors have been associated with the development of AKI. Ritonavir and indinavir (as well as acyclovir, foscarnet, and sulfadiazine) have been associated with reversible AKI thought secondary to crystalluria and intrarenal obstruction (Olyaei et al, 2000). In addition, patients treated with indinavir may present with renal colic because indinavir renal stones can be associated with urinary tract obstruction (Kohan et al, 1999). Newer antiretroviral agents may be toxic to the kidney, and newer forms of AKI related to AIN have recently been described (Cohen et al, 2008). Knowledge of the basic processes involved in the development of ATN is a prerequisite to understanding contemporary therapies directed at limiting renal damage and promoting more rapid renal recovery (Fig. 43–1). In hospital practice, ischemic ATN is the most commonly encountered form of the disease. Therefore the following discussion focuses on ischemic renal injury. (From Brady HR, Brenner BM, Lieberthal W. Acute renal failure. In: Brenner BM, editor. Brenner & Rector’s the kidney. Philadelphia: WB Saunders; 1996. p. 1200–52.) A variety of biochemical changes occur during ischemia and reperfusion that are responsible for the cell dysfunction observed in ATN (Myers et al, 1984; Myers and Moran, 1986). The sentinel biochemical event in renal ischemia is the depletion of adenosine triphosphate (ATP), which is the major energy currency for cellular work. ATP is metabolized to adenosine monophosphate (AMP). During prolonged oxygen deprivation, AMP is further metabolized to the nucleosides adenosine, inosine, and hypoxanthine. These compounds diffuse from the cell, resulting in the loss of the substrate reservoir for ATP synthesis after reperfusion (Brady et al, 1996). Furthermore, hypoxanthine becomes an important substrate in the development of oxygen free radicals during the reperfusion period. Provision of exogenous adenine and inosine decreases cellular injury in experimental renal ischemia (Siegel et al, 1980). ATP depletion results in impaired function of the plasma membrane and intracellular ATPases that are vital to normal cell function. As a consequence of impairment of the Na+/K+ ATPase, cytosolic concentrations of Na+ and K+ are altered and cell swelling results (Alejandro et al, 1995). Dysfunction of the plasma membrane Na+/Ca2+-ATPase and intracellular Ca2+-ATPase leads to high intracellular levels of Ca2+. The increase in intracellular Ca2+ has been associated with multiple aspects of renal cell injury including disruption of the cytoskeleton, activation of Ca2+-dependent phospholipases, acceleration of the conversion of xanthine dehydrogenase to xanthine oxidase (potentiating reperfusion injury), and uncoupling oxidative phosphorylation (Brady et al, 1996). The activation of phospholipases results in damage to the lipid bilayer, which is critical to the normal function of the plasma membrane and intracellular organelles such as mitochondria. Phospholipase activation leads to an accumulation of free fatty acids and lysophospholipids, which are detrimental to vital cellular function, although the mechanism of such action is not clear. Oxidative stress during reperfusion after ischemia is associated with cellular damage (Fig. 43–2). Recall that ATP is metabolized to hypoxanthine. High levels of intracellular Ca2+ activate a calmodulin-dependent protease that converts xanthine dehydrogenase to xanthine oxidase (Brady et al, 1996). The conversion of hypoxanthine to xanthine during reperfusion is the major source of superoxide. This is ultimately metabolized to OH−, which causes cell damage. Finally, the protease calpain is activated and contributes to ischemic renal injury (Edelstein et al, 1997). Calpain regulates membrane channels, kinase activation, and interactions between cytoskeletal proteins. Figure 43–2 The pathophysiology of reperfusion injury. The generation of reactive oxygen species. See text for details. (From Brady HR, Brenner BM, Lieberthal W. Acute renal failure. In: Brenner BM, editor. Brenner & Rector’s the kidney. Philadelphia: WB Saunders; 1996. p. 1200–52.) As the name suggests, ischemic ATN is characterized by renal tubular cell injury. This may be sublethal or lethal. Because obvious necrosis is not a cardinal histopathologic finding in ATN (Racusen et al, 1991), sublethal injury is important. During normal function of the kidney, the medulla operates at the brink of hypoxia due to the countercurrent diffusion of oxygen from the descending to ascending vasa rectae. During prolonged ischemia, medullary hypoxia intensifies and the high metabolic requirement of the nephron structures located in the outer medulla is most sensitive to injury. The S3 portion of the proximal tubule sustains the most severe injury (Witzgall et al, 1994). Other structures that sustain injury in this region include the medullary thick ascending limb (mTAL), which is metabolically active and rich in the energy requiring Na+/K+-ATPase. Sublethal injury to tubular cells leads to aberrations in the cytoskeletal organization of the tubule cells (Molitoris, 1991) (Fig. 43–3). This is manifest as a loss in the cell polarity. The brush border disappears, and there is redistribution of the basolateral Na+/K+-ATPase and integrins (Lieberthal, 1997). As a result, the normal unidirectional transport of salt and water across tubular cells is disrupted and the ability of the renal epithelium to act as a barrier to the free movement of solute and water is lost. The loss of the tight junctions permits backleak of glomerular filtrate, which has been one of the well-established pathophysiologic features of ATN (see Fig. 43–1). In addition to the loss of tight junctions, there is a loss in cell-matrix adhesion (Gailit and Clark, 1993). The redistribution of the integrins disrupts the normal cell adherence to the tubular basement membrane. As a result, abnormal cell–cell adherence develops and contributes to tubular obstruction. The validation of this concept is provided by research demonstrating that an excess of a matrix protein sequence (RGD peptides), which is a critical ligand for the β1 integrin, can ameliorate the reduction in GFR in experimental ATN (Noiri et al, 1994). Conceptually this works because the integrin receptors are saturated by the supplemented matrix protein sequence ligand, making them unavailable for abnormal cell–cell adhesion. This can reduce tubular obstruction and backleak of glomerular filtrate. (From Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med 1996;334:1448–60. Copyright 1996, Massachusetts Medical Society. All rights reserved.) Following sublethal injury, the kidney has a remarkable capacity for repair of normal structure and function. The study of renal recovery from ATN is a relatively new concept with great potential for clinical application. Increased mitotic activity and epithelial regeneration are notable features of ATN (Thadhani et al, 1996). Certain aspects of renal recovery duplicate events in renal development (Witzgall et al, 1994). A number of growth factors play a role in recovery. Epidermal growth factor, insulin growth factor 1, and hepatocyte growth factor have been demonstrated to limit renal injury and accelerate renal recovery in experimental ischemic ATN (Thadhani et al, 1996). RBF is reduced by 50% or more in ischemic ATN, and the perfusion defect is most marked in the outer medulla. The two predominant reasons for this include vasoconstriction and congestion of the medullary vasculature by leukocytes, red cells, and platelets. Ischemic renal injury is marked by intrarenal vasoconstriction as a result of endothelial cell injury. Vasoconstriction results from the imbalance between endothelin (ET) and endothelial-derived nitric oxide (EDNO) (Lieberthal, 1997). Endothelin receptor blockers have been shown to ameliorate ischemic renal damage and improve renal function (Lieberthal, 1997). The endothelial injury sustained in ATN also leads to decreased production of EDNO by constitutive nitric oxide synthase. The decreased EDNO leads directly to vasoconstriction but also permits increased ET production (Lieberthal, 1998). The persistent hemodynamic abnormalities in ATN are also maintained by congestion of the medullary vasculature. Current evidence suggests that ischemic injury results in the release of inflammatory mediators that activate adhesion molecules on leukocytes and upregulates their receptors on the endothelium (Kinsey et al, 2008). Antibodies directed at leukocyte adhesion molecules or their endothelial ligands (i.e., ICAM-1) ameliorate ischemic renal injury (Kelly et al, 1996; Dragun and Haller, 1999). Neutrophils play an important part in the injury cascade. In models where the effects of neutrophils are eliminated by delivering antibodies to certain chemokines, injury is ameliorated (Miura et al, 2001). Interestingly, the tubular epithelium plays an active role in this inflammatory response by generating chemokines that potentiate recruitment of inflammatory cells (Bonventre and Zuk, 2004). These include monocyte chemoattractant protein-1 (MCP-1), interleukin 8 (IL-8), regulated-on activation, normal T cells expressed and secreted (RANTES), and epithelial neutrophil-activating protein 78 (ENA-78). In addition to the role of neutrophils there is evolving evidence that lymphocytes are important in the ischemia/reperfusion injury paradigm. Knockout mice lacking CD4+/CD8+ cell adhesion receptors on T lymphocytes are protected from ischemic injury (Rabb et al, 2000). Also, costimulatory blockade can ameliorate ischemic injury (Takada et al, 1997). Newer agents are being developed to deal with the early inflammatory events in renal ischemia, which may bear practical application, particularly in the setting of renal transplantation. Distinguishing among prerenal, intrinsic renal, and postrenal causes of AKI may prove to be a challenging clinical exercise (Fig. 43–4). A thorough history and physical examination to assess volume status, cardiovascular hemodynamics, potential nephrotoxic insults, and evidence of systemic disease should be undertaken in AKI patients. All interventions and drug therapies surrounding an AKI event should be outlined against the timeline of changes in renal function. Therefore it is critical to know the level of preexisting renal function. One should identify the presence of risk factors known to be associated with AKI such as advanced age; comorbid conditions (heart failure, liver failure, renal insufficiency, diabetes); radiocontrast exposure; therapy with aminoglycoside antibiotics, NSAIDs, or ACE inhibitors; and atheroembolism. In perioperative AKI, critical intraoperative issues to identify include the nature and magnitude of the procedure (open vs. endoscopic), blood loss, hemodynamic stability, integrity of the urinary tract, and intraoperative drug treatment. (Modified from Goldfarb DA, O’Hara JF. Etiology, pathogenesis, and management of peri-operative acute renal failure. American Urological Association Update Series, Lesson 4, vol XX, 2001.) Examination of the urinalysis results is fundamental to the evaluation of the patient with AKI (Perazella and Parikh, 2009). The simple urinalysis may distinguish the cause of AKI among the various possibilities. Table 43–6 highlights the various urinary abnormalities associated with the clinical diagnoses. For example, proteinuria, hematuria, and red blood cell casts are pathognomonic of glomerulonephritis. The classic sediment of ATN includes pigmented (muddy brown) granular casts and renal tubular epithelial cells, which may be seen in nearly 80% of cases of oliguric AKI. Table 43–6 Urine Sediment in Acute Kidney Injury AGN, acute glomerulonephritis; AIN, acute interstitial nephritis; ATN, acute tubular necrosis; RBCs, red blood cells. Determination of urinary chemistry may be helpful in determining the cause of the AKI. The urine sodium, creatinine, and osmolality should be measured. The fractional excretion of sodium or the renal failure index should be calculated (Fig. 43–5). Note that a low fractional excretion of sodium (or renal failure index) may be associated with either prerenal azotemia or AGN (Table 43–7). These entities can be separated clinically by examination of the urinalysis results. Conditions associated with prerenal azotemia have bland urinalysis results, whereas proteinuria, red blood cells, and red blood cell casts are seen with AGN. Other causes of AKI associated with a low fractional excretion of sodium include hepatorenal syndrome and selected types of ATN such as that due to iodinated contrast material, rhabdomyolysis, sepsis, and multisystem organ failure. Both ATN and obstruction may have an increased fractional excretion of sodium or renal failure index. Here again, the urinalysis is crucial in sorting out the differential diagnosis. ATN has a classic sediment with pigmented coarsely granular casts, but the urinalysis results seen in obstruction are often bland with and without microhematuria. Table 43–7 Patterns of Urinary Indices in Acute Kidney Injury AGN, acute glomerulonephritis; ATN, acute tubular necrosis. A variety of imaging modalities have clinical utility in the evaluation of AKI. The most widely used is renal ultrasonography. This noninvasive and readily available study is fairly sensitive for the identification of hydronephrosis, as discussed earlier in the section Postrenal Azotemia. Duplex ultrasonography of the renal artery is useful for the identification of renal artery stenosis (RAS) or thrombosis (Carman et al, 2001). The absence of a Doppler signal from the artery is a noninvasive sign to confirm renal artery thrombosis. Another common imaging study in AKI is the abdominal plain radiograph to identify the presence or location of renal calculi or both. Additionally, the abdominal plain radiograph is particularly helpful to discern the proper position of the stents and drains. The radionuclide renal scan is a useful imaging study in selected clinical circumstances. Tc99-MAG3 is a more useful imaging agent in renal insufficiency than Tc99-DTPA and is able to evaluate both renal flow and function (Taylor, 1999). The renal scan is a simple way to evaluate for renal flow in situations where renal artery thrombosis is a serious consideration such as after partial nephrectomy or renal transplantation. It is especially useful when there is renal insufficiency that prohibits the use of iodinated contrast (Jafri et al, 1988). Urinary extravasation can also be assessed by isotope renography. Management of AKI is based on its cause (Alkhunaizi and Schrier, 1996; DuBose et al, 1997). When AKI is identified as prerenal, correction of the precipitating factors and restoration of renal perfusion usually leads to its resolution. Nephrotoxic drugs should be eliminated when clinically appropriate. Maintaining normal volume status is essential. In the postoperative setting, this implies judicious replacement of crystalloid, colloid, and blood with close monitoring of the central venous pressure. The management of postrenal AKI will depend on its etiology. Any obstruction needs appropriate drainage, and urinary extravasation needs to be controlled. The management of ATN focuses on the prevention of complications and providing an environment that is conducive to renal recovery (Table 43–8). Early consultation with a nephrologist improves the outcome of patients with AKI. Mehta and colleagues (2002) showed that nephrology consultation was delayed in 28% of ICU patients with AKI. Delay in consultation was associated with higher mortality, longer ICU length of stay, and increased number of systems failing at the time of consultation. During the initial evaluation, it is imperative to search for reversible causes such as volume depletion, obstruction, and vascular occlusion. During the initial stages, a trial of parenteral hydration with isotonic fluids may correct AKI secondary to prerenal causes. Thereafter, fluid status should be monitored vigilantly to maintain euvolemia. In a patient with oliguria, special attention must be given to avoiding excessive hydration and volume overload, which might precipitate the need for dialysis. Consideration may be given to using pharmacologic intervention to convert the patient from an oliguric to nonoliguric state. In general, increases in urinary volume make it easier to address problems of volume overload, hyperkalemia, and metabolic acidosis. Increases in urine volume may also provide room for supplemental TPN in the critically ill patient. Historically the morbidity, need for dialysis, and mortality was considered lower in the de novo nonoliguric form of ATN; however, more recent data challenge this notion (Liangos et al, 2005). Table 43–8 Complications of Acute Kidney Injury Pharmacologic intervention to convert oliguric ATN to nonoliguric ATN is a salutary goal for the reasons noted earlier (Townsend and Bagshaw, 2008). Experimental studies on the use of diuretics, dopamine, atrial natriuretic peptide (ANP), and calcium channel blockers (CCBs) that attempt to convert oliguria to nonoliguric AKI have been performed. The applicability of these experimental studies to patients with AKI remains unproved. Uncontrolled studies suggest that patients who respond to mannitol, furosemide, or dopamine with an increased urine output have better outcomes than nonresponders (Cosentino, 1995). The responders may simply have had less severe disease from the outset. Although de novo nonoliguric AKI has been associated with a lower mortality rate, there is little evidence that conversion from an oliguric to a nonoliguric state decreased mortality rate. For the patients with established ATN, therapy with loop diuretics may increase urine output but has little effect on the severity or duration of the AKI (Cosentino et al, 1994). Results of clinical trials involving diuretics, dopamine, ANP, and CCBs for established ATN are reviewed here. Both loop diuretics and mannitol administration have been proved to minimize the degree of renal injury if given at the time of the ischemic insult (Hanley and Davidson, 1981; Schrier et al, 1984; Cosentino, 1995). Both diuretics are capable of inducing a diuresis to wash out obstructive debris and casts. Loop diuretics (e.g., ethacrynic acid, furosemide, and bumetanide) exert their pharmacologic effect in the loop of Henle, causing a large solute diuresis. Additionally, they decrease active NaCl transport in the thick ascending limb of Henle and thereby limit energy requirements in the metabolically active segment, which often bears significant ischemic insult. There are several theoretical reasons why loop diuretics may be of benefit in AKI. Loop diuretics may protect cells of the ascending limb of Henle from hypoxic damage, increase tubular flow to prevent intratubular obstruction, inhibit tubuloglomerular feedback to maintain a favorable GFR, and increase RBF by decreasing renal vascular resistance. The available clinical data do not support an improved outcome in patients who respond to loop diuretics (Shilliday and Allison, 1994). A prospective, randomized placebo-controlled study examining the effect of loop diuretics on renal recovery, dialysis, and death in patients with AKI found no effect (Shilliday et al, 1997). Observational data suggested that diuretic use in critically ill patients with AKI is associated with an increased mortality rate using multivariate analysis and propensity scores (Mehta et al, 2002). A more recent prospective, multicenter, epidemiologic study found that the use of loop diuretics was not associated with higher mortality (Uchino et al, 2004). Therefore it is reasonable to give a trial of a loop diuretic in escalating doses, and if the patient does not respond, the drug should not be readministered because large doses of loop diuretics may be ototoxic and the large infusion volume may cause pulmonary edema. Mannitol, an osmotic diuretic, theoretically ameliorates AKI by flushing intratubular casts, increasing RBF, increasing urine flow, reducing hypoxic cell swelling, protecting mitochondrial function, and scavenging free radicals. Mannitol use continues to be promoted prophylactically in certain high-risk patient groups because of its demonstrated benefit in animal models of AKI (Burke et al, 1983; Shilliday and Allison, 1994). Mannitol has shown some benefit in the clinical setting of AKI (Novick et al, 1983; Weimar et al, 1983; Simmons et al, 2008), particularly when administered prophylactically or within a short time after an ischemic or nephrotoxic insult. The best clinical example of this is its administration before renal artery clamping during partial nephrectomy. However, other studies in humans failed to demonstrate the effectiveness of mannitol in prevention or treatment of ischemic or toxic AKI (Shilliday and Allison, 1994; Baker and Pusey, 2004). Dopamine has selective renal vasodilator properties that cause natriuresis and increased urine output. Low-dose “renal-dose” dopamine (0.4 to 2.0 µg/kg/min) activates dopamine-1 receptors, which induce renal vasodilation and increased RBF. Objective review of controlled studies demonstrates that these benefits remain speculative (Denton et al, 1996; Lassnigg et al, 2000). Bellomo and colleagues (2000) reported on 328 critically ill patients with AKI who were randomly assigned to continuous infusion of placebo or low-dose dopamine (2 µg/kg/min). Peak SCr concentration, requirement for dialysis, length of hospital stay, and mortality rate did not differ between the two groups. Also, prophylactic use of low-dose dopamine in patients undergoing coronary artery bypass surgery has not been shown to be effective in preventing the development of renal impairment in these patients (Woo et al, 2002). Lauschke and colleagues (2006) recently reported on the effects of renal-dose dopamine on renal perfusion as measured by resistive indices obtained by ultrasonography. Although renal perfusion indeed increased in subjects with no AKI, renal vascular resistance actually increased in those with AKI, a finding that was more marked in older patients (Lauschke et al, 2006). It is also important to note that the use of dopamine has been associated with serious cardiac, vascular, and metabolic complications in the critically ill and therefore should be used with caution (Polascik et al, 1999). Other than dopamine, the literature provides little guidance on the effects of other vasoactive agents on the kidney and thus a need for large randomized, controlled studies to clarify this issue (Baker and Pusey, 2004). Fenoldopam is a selective dopamine receptor-1 agonist (DA-1) that causes DA-1 receptor-mediated vasodilation and does not stimulate DA-2 or α- or β-adrenergic receptors (Baker and Pusey, 2004). Fenoldopam reduces renal vascular resistance and increases RBF and fractional excretion of sodium and free water clearance in studies in normal volunteers and hypertensive patients (Mathur et al, 1999). A few studies in animal models (Singer and Epstein, 1998; Halpenny et al, 2001) are consistent with the notion that DA-1 agonists may be useful in preventing or treating AKI. Some clinical studies have been encouraging in the treatment (Stone et al, 2003) and prevention (Kini et al, 2002) of contrast-induced nephropathy and when used perioperatively in cardiovascular surgery (Halpenny et al, 2001). A recent multicenter trial, however, did not find any protective benefit of the selective dopamine-1 agonist (fenoldopam mesylate) in the prevention of contrast-induced renal dysfunction in an at-risk population (Landoni, 2007). One study showed when fenoldopam was used prophylactically in patients undergoing aortic surgery, its use was associated with improvement in renal function, reductions in dialysis requirements, length of hospital stay, and mortality (Gilbert et al, 2001). A meta-analysis of all randomized controlled trials using fenoldopam to prevent AKI, which included 1290 patients from 16 different studies, suggests that the use of this drug may be beneficial in reducing the need for renal replacement therapy, shortening ICU stay, and perhaps improving mortality (Landoni et al, 2007); however, because these studies were small, defining fenoldopam’s role in clinical situations is difficult without a large-scale randomized controlled trial. In some animal studies the effects of ischemic AKI could be reversed with the use of an intrarenal arterial infusion of ANP (Nakamoto et al, 1987; Shaw et al, 1987), but other experimental studies have been contradictory. The proposed mechanism by which this occurs is the vasodilatory action of ANP. In experimental models of established ATN, the combination of ANP with dopamine to prevent systemic hypotension resulted in a rise in GFR induced by arteriolar vasodilatation. The treated animals also exhibited less tubular necrosis and fewer casts, suggesting tubular recovery was promoted. The efficacy of ANP in established ATN in humans has been evaluated in two trials (Rahman et al, 1994). First was a small, randomized trial suggesting a benefit of ANP therapy. More recently, a prospective, multicenter, randomized study with ANP in patients with ATN (oliguric and nonoliguric) has been reported. Overall, there was no established benefit on morbidity and mortality with ANP. However, in a subset of patients with oliguric ATN, clinical improvement was seen with ANP infusion. Further studies in such a select population did not demonstrate benefit (Lewis et al, 2000). ANP is still considered an investigational drug, and its role in the management of patients with ATN remains uncertain. Experimental studies suggest the possibility of accelerating the rate of tubular regeneration in ATN by administration of growth factors such as insulin-like growth factor-1 (IGF-1) (Miller et al, 1992) and epidermal growth factor (EGF) (Hammerman, 1999). The recovery process involves both tubular cell regeneration and differentiation into mature functioning cells. The applicability of these observations to human disease remains to be determined. A report of a randomized trial of AKI patients of less than 7 days’ duration found no benefit in patients treated with IGF-1 (Hirschberg et al, 1999). In a large, randomized, double-blind, placebo-controlled trial in intensive care units in 20 teaching hospitals involving 72 patients, it was shown that IGF-1 does not accelerate the recovery of renal function in AKI patients with substantial comorbidities (Hirschberg et al, 1999). Similarly, there is no role for thyroxine in modifying the course and outcome in AKI; in fact, it could have a negative effect on outcome through prolonged suppression of TSH (Acker et al, 2000). Therefore on the basis of current evidence, there is no role for the use of growth factors to treat AKI. Calcium channel blockers inhibit voltage-gated calcium entry into cells and are reported to reverse vascular constriction, increase GFR, and improve renal plasma flow (Epstein, 1993). A few limited animal studies in experimental AKI generally support a protective benefit of calcium channel blockers. The clinical benefit of calcium channel blockers most widely studied has been the effect on graft function in renal transplant recipients (Alkhunaizi and Schrier, 1996). Once the clinical diagnosis of ATN is made, conservative medical management is in order (Table 43–9). This would include attempts to minimize further renal parenchymal injury, ensure provision of nutrition, maintain the metabolic balance, and promote recovery of renal function. Table 43–9 Conservative Medical Management of Acute Kidney Injury Optimizing the patient’s volume status is imperative, particularly in patients with oliguric AKI (DiBona, 1994). If such patients are administered large volumes of IV fluid or allowed free access to oral fluids, they are at risk for developing fluid overload. In the oliguric patient, fluids should be restricted to total output plus insensible losses. If required, pharmacologic intervention with loop diuretics may promote increases in urinary volume. Providing adequate nutrition is important for the recovery of the critically ill patient with AKI (Fiaccadori et al, 2008). Preexisting or hospital-acquired malnutrition is an important factor contributing to high mortality seen in patients with AKI (Druml, 1998; Kriz et al, 1998). AKI not only affects water, electrolyte, and acid-base metabolism but induces specific alterations in protein and amino acid, carbohydrate, and lipid metabolism (Druml, 1992). The metabolic alterations in AKI patients are determined not only by acute loss of renal function but also by the underlying disease process (i.e., sepsis, trauma, or multiple organ failure) and by the type and intensity of renal replacement therapy (RRT) (Druml, 1992, 2005). The hallmark of metabolic alterations in AKI is activation of protein catabolism with excessive release of amino acids from skeletal muscle and sustained negative nitrogen balance (Druml, 1998; Price et al, 1998). Hepatic extraction of amino acids from the circulation, gluconeogenesis, and ureagenesis are all increased. Several additional catabolic factors (secretion of catabolic hormones, hyperparathyroidism, suppression and decreased sensitivities to growth factors, and release of inflammatory mediators) are operative in AKI. All of these factors mediate protein breakdown (Cianciaruso et al, 1991). Frequently, AKI is associated with hyperglycemia caused by insulin resistance (Klouche and Beraud, 1998). When plasma insulin concentration is elevated, maximal insulin-stimulating glucose uptake by skeletal muscle is decreased by 50%. AKI is also associated with accelerated hepatic gluconeogenesis, mainly from conversion of amino acids released during protein catabolism, which cannot be suppressed by exogenous glucose infusions (Druml, 1992). The triglyceride content of plasma lipoprotein is increased in AKI, whereas total cholesterol and high-density lipoprotein (HDL) cholesterol are decreased (Schneeweiss et al, 1990). The major cause of lipid abnormalities in AKI is impairment of lipolysis. RRTs themselves may have significant metabolic and nutritional consequences. Appropriate nutritional therapy in patients with AKI may be beneficial in promoting recovery (Fiaccadori et al, 2008). Caloric intake should be maintained, and carbohydrate intake should be at least 100 g/day to minimize ketosis and endogenous protein catabolism. A moderate protein intake of about 1 to 1.8 g/kg by weight per day may be required to maintain positive nitrogen balance. Higher protein intakes of up to 2.5 g/kg/day have been necessary to improve nitrogen balance in critically ill AKI patients on continuous dialysis, although no survival advantage was noted. Hence it should be stressed that a low-protein intake (<0.5 g/kg/day) may be unnecessary and protein intake should not be severely restricted in AKI to limit the need for dialysis. In terms of types of amino acids used, diet or solutions including both essential and nonessential amino acids in standard proportions are recommended. Dietary phosphorus, potassium, and sodium chloride may be restricted. In the critically ill patient, nutritional support via TPN or enteral feedings should be considered. Prior studies demonstrate that provision of adequate nutrition to the AKI population may improve survival (Chertow et al, 2000). Patients with AKI are candidates to develop significant electrolyte abnormalities such as hyperkalemia, metabolic acidosis, hyperphosphatemia, and hypocalcemia. These problems may be minimized by the prophylactic institution of a low-potassium diet accompanied by fluid restriction and oral phosphate binders. The initiation of dialysis for patients with AKI is usually precipitated by one or more of the specific indications. There is still debate on whether patient morbidity and mortality might be improved by early, more intensive dialysis to keep the BUN less than 80 to 100 mg/dL. A recent study from a multicenter trial suggested that in those patients who developed AKI requiring dialysis, patients with BUN levels lower than 76 mg/dL had a better prognosis than those with higher levels, suggesting that initiation of treatment at earlier time points may be beneficial (Liu et al, 2006
Acute Kidney Injury
Definition
Epidemiology and Classification of Acute Kidney Injury
Prerenal Azotemia
Postrenal Azotemia
Intrinsic Renal Disease
Acute Glomerulonephritis
Multisystem Diseases
Superimposed on Primary Glomerular Disease
Infectious Diseases
Drugs and Toxic Agents
Idiopathic
Acute Interstitial Nephritis
Acute Tubular Necrosis
Incidence and Etiology
Antibiotics
Anesthetic Agents
Contrast Media
Antiulcer Regimens
Diuretics
Chemotherapeutic and Immunosuppressive Agents
Analgesics
HIV Protease Inhibitors
Organic Solvents
Heavy Metals and Poisons
Chemicals*
Recreational Drugs†
Miscellaneous
Pigment Nephropathy
Intrarenal Crystal Deposition
Tumor-Specific Syndromes
Pathophysiology
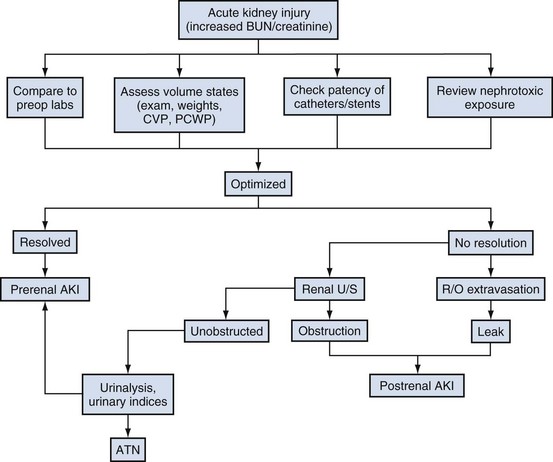
Clinical Approach to the Differential Diagnosis of Acute Kidney Injury
General
SEDIMENT FINDINGS
DIAGNOSIS
Normal
Prerenal/obstruction
RBC casts, RBCs
AGN/vasculitis
Eosinophils
AIN
Pigmented granular casts
ATN
PRERENAL/AGN
ATN/OBSTRUCTION
Urinary [Na+] mEq/L
<20
>40
Urine: plasma creatinine
>30
<20
Renal failure index
<1
>1
FENa
<1
>1
Urinary osmolality
>500
<400
Imaging
Management of Acute Kidney Injury
Fluid Overload
Electrolyte Disturbances
Uremic Signs and Symptoms
Neurologic
Cardiac
Pulmonary
Hematologic
Immunologic
Pharmacologic Intervention
Conservative Management
Fluid Balance
Electrolytes and Acid-Base Balance
Uremia and Nutrition
Drugs
Dialytic Interventions
General
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Prostate Cancer Tumor Markers
Prostate Cancer Tumor Markers
 Core Principles of Perioperative Management in Children
Core Principles of Perioperative Management in Children
 Neuropathic Dysfunction of the Lower Urinary Tract
Neuropathic Dysfunction of the Lower Urinary Tract
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Etiology, Pathogenesis, and Management of Renal Failure
B Cardiac causes: Primary decrease in cardiac output
1. Acute disorders: myocardial infarction, arrhythmias, malignant hypertension, tamponade, endocarditis