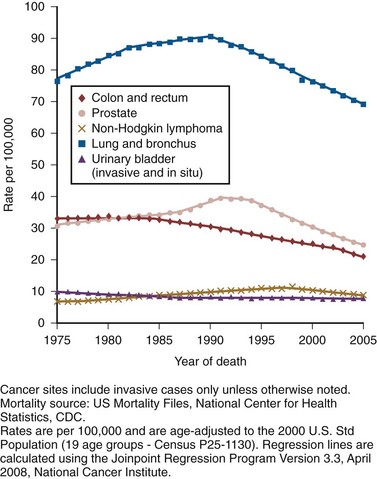
Robert Abouassaly, MD, MSc, Ian M. Thompson, Jr., MD, Elizabeth A. Platz, ScD, MPH, Eric A. Klein, MD Prostate cancer has been the most common noncutaneous malignancy in U.S. men since 1984, now accounting for one quarter of all such cancers (American Cancer Society, 2008). The estimated lifetime risk of disease is 16.72%, with a lifetime risk of death at 2.57%. Prostate cancer incidence varies by race/ethnicity, with African-Americans at highest risk (Table 95–1). The incidence of prostate cancer peaked in 1992 (approximately 5 years after introduction of the prostate-specific antigen [PSA] screening test), fell precipitously until 1995, increased slowly until 1995 at a slope similar to that observed prior to the PSA era, and has decreased again in recent years (Fig. 95–1). The years in which peak rates were observed can be differentiated by race: 1992 for whites (237.8 per 100,000 men) and 1993 for African-Americans in (343.1 per 100,000). The decrease in incidence between 1992 and 1995 has been attributed to the “cull effect” of identifying previously unknown cancers in the population by the use of the PSA test, followed by a return to baseline where fewer cases were detected in previously screened individuals (Stephenson et al, 1996). For 2008, the American Cancer Society estimated 186,320 new cases of prostate cancer in the United States (American Cancer Society, 2008). Table 95–1 Prostate Cancer Incidence and Mortality by Race/Ethnicity, United States, 2000–2004 * Per 100,000, age adjusted to the 2000 U.S. standard population. Data from American Cancer Society. Cancer facts and figures 2008. <http://www.cancer.org/acs/groups/content/@nho/documents/document/2008cafffinalsecuredpdf.pdf>; 2008 [accessed 06.04.11]. Figure 95–1 Age-adjusted cancer incidence rates for men, United States, 1975-2005. (From Surveillance, Epidemiology, and End Results Program, 1975-2005, Division of Cancer Control and Population Sciences, National Cancer Institute, 2008.) Prostate cancer mortality rates in the United States rose slowly between 1973 and 1990 (Fig. 95–2). This may have resulted from a gradual increase in the number of biologically lethal cancers or a decreasing use or effectiveness of therapy during this interval. In the early 1990s, an abrupt rise in mortality was observed. This increase may have been caused by an increase in attribution bias occurring when the National Center for Health Statistics made a change from manual to automated methods for assignment of cause of death (Feuer et al, 1999). Subsequent to 1991, the peak mortality year, steady declines in prostate cancer mortality were reported. The magnitude of this decline is nearly 2.5 times larger than the increase in mortality seen as a result of attribution bias; thus it seems likely that the declines in prostate cancer mortality in the United States since 1991 are real and clinically significant (Stephenson, 2005). In 2008, the American Cancer Society estimated 28,660 prostate cancer–related deaths in the United States, for an approximate annual rate of 23.3 per 100,000 population, representing a 41% decrease from the peak in 1991 (American Cancer Society, 2008). Furthermore, the mortality rate for prostate cancer in white men in the United States has declined to a level lower than that observed prior to the introduction of PSA-based screening in 1987 (Tarone et al, 2000). Figure 95–2 Age-adjusted cancer death rates for men, United States, 1975-2005. (From Surveillance, Epidemiology, and End Results Program, 1975-2001, Division of Cancer Control and Population Sciences, National Cancer Institute, 2008.) The observed decline in mortality since 1991 is unlikely to be explained by PSA screening alone. Based on our current beliefs concerning lead time, the decline would have occurred too soon after the initiation of screening (Etzioni et al, 1999). One hypothesis is that it is the result of the more aggressive treatment of prostate cancer that began in the 1980s (Walsh, 2000). Rates of both radical prostatectomy (RP) and radiation therapy rose steadily through the 1980s (pre-PSA era), whereas hormone therapy and no-treatment rates remained stable (Stephenson, 2005). Outcomes for patients treated in the 1980s should be reflected in the mortality data of the 1990s, while outcomes for patients treated in the PSA era (the 1990s) have had less time to affect recent mortality data. Given the long natural history of low-stage cancers detected in the PSA era, their treatment would not be expected to have a substantial effect on mortality statistics for another 5 to 10 years. Additional observation time is necessary to determine if screening, PSA-induced stage migration, and more aggressive use of therapy have contributed to declining mortality. Although anthropologists accept that there are subtle biologic differences among populations, commonly used categories, such as African-American, white, and Hispanic, are social and cultural descriptors that have no defined biologic basis. Observed disease-related differences between groups defined in this fashion may therefore be more reflective of common environmental exposure, diet, lifestyle, and attitudes toward health care than of differences in genetic structure or function. Recognizing these caveats, it is noteworthy that African-American men have the highest reported incidence of prostate cancer in the world, with a relative incidence of 1.6 compared with white men in the United States (American Cancer Society, 2008). Although African-Americans have experienced a greater decline in mortality than white men since the early 1990s, their death rates remain more than 2.4 times higher than whites. A recent meta-analysis quantifies this difference, demonstrating a higher risk of biochemical recurrence (risk ratio = 1.34, 95% confidence interval [CI] = 1.23 to 1.46), prostate cancer–specific death (risk ratio = 1.29, 95% CI = 1.13 to 1.47), and all-cause mortality (risk ratio = 1.35, 95% CI = 1.23 to 1.48) (Evans et al, 2008). These differences were not fully explained by differences in comorbidity, PSA screening, or access to health care. One study that analyzed Medicare data found shorter survival intervals for African-Americans: 1.8 years shorter with localized disease treated by RP, 0.7 years shorter after radiation, and 1 year shorter in those choosing watchful waiting, findings that persist after adjusting for other covariates, including education and income levels (Godley et al, 2003). Many biologic, environmental, and social hypotheses have been advanced to explain these differences, ranging from postulated differences in genetic predisposition; differences in mechanisms of tumor initiation, promotion, and/or progression; higher fat diets, higher serum testosterone levels, or higher body mass index; structural, financial, and cultural barriers to screening, early detection, and aggressive therapy; and physician bias. Differences in screening rates between whites and African-Americans may play a role in explaining the differences in mortality, although the 2005 National Health Interview Survey demonstrated that among men ages 40 to 49 years, African-American men were more likely to have had a PSA test than white men, and there were no significant differences in PSA screening rates by race/ethnicity in men ages 50 to 79 years (Ross et al, 2008). Studies have consistently shown that African-American men are more likely to receive androgen-deprivation therapy, expectant management or external beam radiation therapy, and are less likely to undergo radical prostatectomy compared with white men (Underwood et al, 2005). Even among those treated with “watchful waiting,” African- American men may receive less intensive follow-up (Shavers et al, 2004). There are currently no data that clearly indicate any of these hypotheses are the determinants of the observed differences in incidence or mortality, and it seems likely that the source of the disparity is multifactorial. Recent observations suggest that the incidence of organ-confined disease at diagnosis among African-Americans is increasing, that the disparity in mortality is lessening in the PSA era, and that those with organ-confined disease can be cured at a high rate regardless of race (Powell et al, 2004; American Cancer Society, 2008). The incidence of prostate cancer in other ethnic groups is lower than that of whites and African-Americans. Comparative data for prostate cancer–related incidence and mortality are now available for these groups (see Table 95–1). Incidence rates show that prostate cancer is the fifth most common malignancy worldwide and the second most common in men (Parkin et al, 2005). Prostate cancer makes up 11.7% of new cancer cases overall, 19% in developed countries, and 5.3% in developing countries. Its incidence varies widely between countries and ethnic populations, with disease rates differing by more than 100-fold. The lowest yearly incidence rates occur in Asia (1.9 cases per 100,000 in China) and the highest in North America and Scandinavia, especially in African-Americans (249 cases per 100,000) (Parkin et al, 2005; American Cancer Society, 2008). As in the United States, prostate cancer incidence has increased in many countries since the early 1990s, with the largest increases in high-risk countries (Hsing et al, 2000). Although much of the increase can be correlated with the introduction of PSA, some of the increase predates screening and is perhaps partly due to the increasing diagnosis of latent cancers following transurethral resection of the prostate (Potosky et al, 1990; Oliver et al, 2001). Large increases have also been noted in low-risk countries between 1973 and 1992 (e.g., 104% among Chinese in Singapore and 84% in Miyagi, Japan) (Hsing et al, 2000). Mortality also varies widely among countries, being highest in the Caribbean (28 per 100,000 per year) and lowest in Southeast Asia, China, and North Africa (<5 per 100,000 per year) (Parkin et al, 2005). Mortality rates increased slowly for most countries between 1985 and 1995 (Quinn and Babb, 2002). The CONCORD study, a worldwide population-based analysis of cancer survival in five continents, recently analyzed international differences in survival for breast, colorectal, and prostate cancer (Coleman et al, 2008). Using data from cancer registries, age-standardized 5-year survival rates were found to vary greatly, ranging from 80% or higher in the United States (92%), Australia, and Canada, to less than 40% in Denmark, Poland, and Algeria. There are multiple potential explanations for the worldwide and ethnic variations in prostate cancer incidence and mortality. Access to and quality of health care, the accuracy of cancer registries, and the penetrance of PSA screening all affect how rates of disease are reported. Before reliable data were available from African countries, rates of prostate cancer in Africa were thought to be much the same as those in Asia. However, in Uganda and Nigeria, prostate cancer is very common, and in Nigeria it is the most common cancer in men (Gronberg, 2003). Interestingly, there has been a more marked decline in prostate cancer mortality in countries with a higher uptake of PSA screening when compared with countries where routine screening is not widely performed (Collin et al, 2008). Possible explanations for the observed mortality differences include international variability in treatment approaches and bias related to the misattribution of cause of death. Environment also plays an important role in modulating prostate cancer risk around the world. Japanese and Chinese men in the United States have a higher risk of developing and dying from prostate cancer than do their relatives in Japan and China (Muir et al, 1991; Shimizu et al, 1991). Likewise, prostate cancer incidence and mortality have increased in Japan as the country has become more Westernized (Hsing et al, 2000). It is important to note, however, that Asian-Americans have a lower prostate cancer incidence than white or African-American men, indicating that genetics still plays a role in determining prostate cancer predisposition. Prostate cancer is rarely diagnosed in men younger than 50 years old, accounting for only 2% of all cases (Jani et al, 2008). The median age at diagnosis is 68 years, with 63% diagnosed after age 65 (Ries et al, 2011). At 85 years of age, the cumulative risk of clinically diagnosed prostate cancer ranges from 0.5% to 20% worldwide, despite autopsy evidence of microscopic lesions in approximately 30% of men in the fourth decade, 50% of men in the sixth decade, and more than 75% of men older than 85 years (Sakr et al, 1993; Gronberg, 2003). PSA-based screening has induced an important age migration effect; the incidence of prostate cancer in men 50 to 59 years of age has increased by 50% between 1989 and 1992 (Hankey et al, 1999), with important implications for deciding on the need for, type of, and complications after therapy. In addition to changes in prostate cancer incidence and mortality over the last several decades, there has been a substantial shift to more favorable stage at presentation in men with newly diagnosed disease. This clinical stage migration is largely if not exclusively accounted for by PSA screening (Catalona et al, 1993; Mettlin et al, 1993). Since the introduction of PSA testing, the incidence of local-regional disease has increased, whereas the incidence of metastatic disease has decreased (Newcomer et al, 1997). Nonpalpable cancers (American Joint Committee on Cancer [AJCC] clinical stage T1c) now account for 60% to 75% of newly diagnosed disease (Derweesh et al, 2004; Gallina et al, 2008). Clinical stage migration has also been associated with improvements in 5- and 10-year disease-specific survival, which for all stages combined now are 99% and 91%, respectively (American Cancer Society, 2008). The use of PSA has also resulted in a substantial downward pathologic stage migration as evidenced by an increase in the proportion of patients with organ-confined disease (Catalona et al, 1993; Jhaveri et al, 1999; Derweesh et al, 2004😉 and a decrease in the proportion with seminal vesicle involvement (Gallina et al, 2008). These observations have been consistent across the United States and Europe (Noldus et al, 2000; Gallina et al, 2008). Since 1995, however, a slowing in this trend has been observed, suggesting a diminishing effect of PSA screening on pathologic stage migration (Fig. 95–3) (Dong et al, 2007b). The improvement in pathologic stage has been seen for clinical stages T1-T3 tumors and all tumor grades (Fig. 95–4) and has resulted in improved cancer-specific survival after external radiation or surgery for patients treated late in the PSA era (Jhaveri et al, 1999; Derweesh et al, 2004; Kupelian et al; 2005). Recently, the results of two large randomized trials assessing the effect of PSA screening on prostate cancer mortality were published. The first examined results from the Prostate, Lung, Colorectal, and Ovarian (PLCO) cancer screening trial and reported on 76,693 men aged 55 to 74 years at 10 U.S. centers receiving either annual screening or usual care (Andriole et al, 2009). After 7 years of follow-up, no difference in prostate cancer mortality was detected between the groups, with an incidence of 2.0 deaths per 10,000 person-years (50 deaths) in the screening group and 1.7 deaths per 10,000 person-years (44 deaths) in the control group (rate ratio = 1.13, 95% CI = 1.16 to 1.29). However, the trial has been criticized for its relatively short follow-up, given the long natural history for prostate cancer and because more than half the men in the control arm actually had a PSA test done outside of the trial. The second trial, the European Randomized Study of Screening for Prostate Cancer (ERSPC), included 162,243 men between the ages of 55 and 69 years randomized to either PSA screening every 4 years or no screening (Schroder et al, 2009). After a median follow-up of 9 years, screening reduced the rate of death from prostate cancer by 20% (rate ratio = 0.80, 95% CI = 0.65 to 0.98). However, they estimated that to prevent one prostate cancer death, one would need to screen 1410 men (95% CI = 1142 to 1721), and treat 48 additional cases of prostate cancer. The results of these trials highlight two important points. First, overall, prostate cancer is an indolent disease with a very low cause-specific death rate and will only impact life expectancy in a minority of men. Second, the morbidity of treatment needs to be strongly considered when evaluating population screening. Prior to the release of the above-mentioned studies, an evidence update for the U.S. Preventative Task Force on the benefits and harms of PSA screening concluded that false-positive PSA test results cause psychologic adverse effects for up to 1 year and that the benefits of screening remain uncertain (Lin et al, 2008). On the other hand, in 2009 the American Urological Association (AUA) released a Best Practice Statement suggesting that early detection of and risk assessment for prostate cancer should continue to be offered to asymptomatic men 40 years of age or older who have a life expectancy of at least 10 years (American Urological Association, 2009). Key Points Epidemiology Ample epidemiologic evidence suggests that prostate cancer has both a familial and genetic component. The first reports of a familial clustering were published in the mid-20th century and suggested that the risk of developing prostate cancer was higher in those with an affected first-degree relative (Woolf, 1960). Subsequent case control and cohort studies have confirmed this observation (Eeles et al, 1997). Twin studies have also suggested a genetic component, with higher rates of concordance for monozygotic than dizygotic brothers (Gronberg et al, 1994; Ahlbom et al, 1997; Page et al, 1997). Results of a meta-analysis demonstrate that relative risk increases according to the number of affected family members, their degree of relatedness, and the age at which they were affected (Table 95–2) (Zeegers et al, 2003). A consistent finding is the increased risk when a brother is affected compared with when a father is affected. This is consistent with either X-linked or recessive genetic components (Monroe et al, 1995). However, the bias associated with the fact that brothers of affected men are more likely to seek prostate cancer screening may also explain the differential association. Table 95–2 Family History and Risk of Prostate Cancer Data derived from meta-analysis assessing risk of prostate cancer for relatives of patients with prostate carcinoma (Zeegers, Jellema A, Ostrer H. Empiric risk of prostate carcinoma for relatives of patients with prostate carcinoma: a meta-analysis. Cancer 2003;97:1894–03). For investigative purposes, prostate cancer may be conveniently divided into three phenotypes: sporadic, familial, and hereditary. Sporadic cancers occur in individuals with a negative family history. Familial prostate cancer is defined as cancer in a man with more than one affected relative. Hereditary prostate cancer is a subset of the familial form and has been operationally defined as nuclear families with more than three affected members, prostate cancer in three successive generations, or two affected individuals diagnosed with cancer before age 55 (Carter et al, 1993). Although most prostate cancer is likely to be polygenic in origin, the existence of true hereditary prostate cancer is suggested by three epidemiologic observations: (1) relatives of patients < 55 years old are at higher risk of getting prostate cancer than those with older affected relatives; (2) there is stronger familial clustering in families with early onset prostate cancer; and (3) the number of affected family members and their age at onset are the most important determinants of risk among relatives. Sporadic cancers account for about 85% of all prostate cancers and about 15% are familial and/or hereditary. Hereditary prostate cancer accounts for 43% of early onset disease (55 years of age or younger) but only 9% of all cancers that occur by 85 years of age (Carter et al, 1992). A number of studies have suggested a familial coaggregation of prostate cancer with breast cancer (Thiessen, 1974; Tulinius et al, 1992; Goldgar et al, 1994). The two major susceptibility genes in breast cancer, BRCA1 (17q21) and BRCA2 (13q12), have also been studied in prostate cancer. Two large epidemiologic studies have estimated the risk of prostate cancer in men who are carriers of specific BRCA1 founder mutations to be double that of noncarriers, with a cumulative risk of 30% by 80 years of age (Struewing et al, 1997; Warner et al, 1999). Moreover, the BRCA2 gene has provided consistent association with an increased risk of prostate cancer, estimated at 5- to 7-fold, and may play a more important role in early onset disease (Breast Cancer Linkage Consortium, 1999; Edwards et al, 2003). However, due to their rarity, these mutations only contribute to a small fraction of hereditary prostate cancer cases. Linkage studies have identified a number of candidate prostate cancer susceptibility genes, including RNaseL (hereditary prostate cancer-1 [HPC1] region, 1q23-25) (Carpten et al, 2002), ELAC2 (HPC2 region, 17p) (Tavtigian et al, 2001), and MSR1 (8p22-23) (Xu et al, 2002). Segregation studies have also suggested the existence of prostate cancer susceptibility loci on chromosomes 1q42.2-43 (PCAP) (Berthon et al, 1998), 1p36 (CAPB, also linked to brain tumors) (Gibbs et al, 1999), and Xq27-28 (Xu et al, 1998), but the gene or genes linked to these regions have not been identified. Many other genetic loci and prostate cancer susceptibility genes have been more recently identified, and the list is continually growing (Online Mendelian Inheritance in Man, 2009). More recently, genome-wide association studies (GWAS) have emerged as a new approach to indentify alleles associated with prostate cancer without prior knowledge of position or function (Wellcome Trust Case Control Consortium, 2007). Genotype frequencies are compared among cases and controls at DNA sequence variations called single nucleotide polymorphisms (SNPs). Using this technique, studies have demonstrated SNPs on 8q24 and 17q, as well as on chromosomes 3, 6, 7, 10, 11, 19, and X, to be associated with prostate cancer risk (Amundadottir et al, 2006; Gudmundsson et al, 2007; Eeles et al, 2008). A worldwide consortium of 13 groups was recently formed to compile the large series necessary to provide accurate estimates of the risks associated with genetic variants and to evaluate the combined association of these variants (Kote-Jarai et al, 2008). The combined risks associated with each pair of SNPs were found to be consistent with a multiplicative risk model. In total, they determined that the 15 susceptibility variants studied only explained 16% of the familial risk of prostate cancer. This indicates that, to date, only a small piece of the prostate cancer genetic puzzle has been discovered, and with the continued improvement in genetic technology, it is likely that the number of known susceptibility genes will increase. A commercially available genetic test, which assesses nine genetic variants associated with prostate cancer risk, is currently offered to the general public (deCODE Genetics, Reykjavík, Iceland). Although the test is being marketed as a preventative tool, its clinical utility in the general population requires further study. Of the known susceptibility genes, HPC1 is the best characterized. The existence of HPC1 was suggested by a genome-wide scan of families with hereditary prostate cancer (Smith et al, 1996) and later confirmed by linkage studies (Cooney et al, 1997; Eeles et al, 1998). A recent genome-wide association screen also identified an association between this locus and sporadic prostate cancer (Nam et al, 2008). HPC1 was mapped to the human gene that encodes for the antiviral and pro-apoptotic enzyme RNaseL (Carpten et al, 2002). RNaseL is part of the innate immune system that responds to a pathogen-associated molecular pattern (PAMP) to induce degradation of viral and cellular RNAs, thereby blocking viral infections (Klein and Silverman, 2008). It is the terminal enzyme of the 2-5A synthetase family, an RNA degradation pathway that plays an important role in mediating the biologic effects of interferons, especially in response to viral infection. 2-5A binds with high affinity to RNaseL, converting it from its inactive form as a monomer to a potent dimer that degrades single-stranded RNA, thus preventing viral replication, interfering with protein synthesis, and causing caspase-mediated apoptosis (Fig. 95–5) (Silverman, 2003). RNaseL knockout mice are more prone to viral infections (Silverman, 2003). (Reprinted with permission, Cleveland Clinic Center for Medical Art & Photography © 2005-2011. All Rights Reserved.) A variety of inactivating and missense mutations of RNaseL have been identified in families with hereditary prostate cancer (Xiang et al, 2003). Of these, SNP R462Q, resulting from an arginine to glutamine substitution, has been shown to be associated with an increased risk of prostate cancer (Casey et al, 2002); men and cell lines with this allelic variant have been shown to have reduced RNaseL activity leading to deficient apoptosis (Carpten et al, 2002; Xiang et al, 2003; Malathi et al, 2004), presumed to lead to an accumulation of genetic defects ultimately resulting in cancer formation. The epidemiologic data suggest that HPC1 is a rare autosomal dominant gene that has high penetrance, meaning that while it does not account for many prostate cancers, an individual carrier is highly likely to be affected by prostate cancer. Although cancers with specific mutations of the HPC1/RNaseL gene have been reported to present with higher-grade and more advanced stages (Gronberg et al, 1997; Goode et al, 2001; Rennert et al, 2005), other mutations have not shown such an association (Larson et al, 2008). Key Points Risk Factors It is estimated that infections cause almost 20% of all cancers worldwide (American Cancer Society, 2008). Chronic inflammation leading to cellular hyperproliferation to replace damaged tissue contributes to the development of infection-associated cancers of the colon, esophagus, stomach, bladder, and liver (Coussens and Werb, 2002; De Marzo et al, 2007). Accumulating epidemiologic, histologic, and genetic evidence suggests that a similar process may underlie the development of prostate cancer. Some evidence suggests that prostate cancer may have an infectious etiology. Two meta-analyses examining 34 case-control studies reported statistically significant associations of prostate cancer with a history of sexually transmitted infection (RR = 1.4) or prostatitis (OR = 1.57) (Dennis et al, 2002b; Dennis and Dawson, 2002). Supportive evidence is provided by studies demonstrating positive associations of antibodies against syphilis, human papillomavirus (HPV), and human herpesvirus-8 (HHV-8) with prostate cancer (De Marzo et al, 2007). Case-only or case-control studies have also reported higher plasma concentrations of acute-phase reactants and proinflammatory cytokines in men with prostate cancer (De Marzo et al, 2007). Two studies have demonstrated evidence of viral pathogens in human prostate tissue, including HPV, human herpes simplex virus type 2 (HSV2), cytomegalovirus (CMV), and HHV-8 (Strickler and Goedert, 2001; Zambrano et al, 2002; Samanta et al, 2003). However, recent studies assessing the association between infection and prostate cancer have shown mixed results. In the Health Professionals Follow-up Study (Sutcliffe et al, 2006), a prospective study of 51,529 American male health professionals aged 40 to 75 years, no association was found between a self-reported history of gonorrhea or syphilis and prostate cancer, although the incidence of sexually transmitted infections was very low in this population. In addition, no overall correlation between prostate cancer and clinical prostatitis was found. Similarly in this cohort, no associations were observed between Chlamydia trachomatis, HPV-16, HPV-18, and HPV-33 antibody seropositivity and prostate cancer (Sutcliffe et al, 2007a). Conversely, in a small case-control study in African-American men (Sarma et al, 2006), a history of gonorrhea or prostatitis increased the odds of prostate cancer by 1.78-fold (95% CI = 1.13 to 2.79) and 4.93-fold (95% CI = 2.79 to 8.74), respectively, even after adjusting for potential confounders. Furthermore, men reporting 25 or more sexual partners had 2.80 times the odds of being diagnosed with cancer (95% CI = 1.29 to 6.09) compared with men with five or fewer partners. Inflammatory infiltrates and the histologic lesion called proliferative inflammatory atrophy (PIA) are frequent in clinical prostate specimens (De Marzo et al, 1999). PIA is a spectrum of lesions characterized by epithelial atrophy, low apoptotic index, and an increased proliferative index usually associated with inflammatory infiltrates (Putzi and De Marzo, 2000). Inflammation in PIA may include mononuclear infiltrates in the stroma and macrophages and/or neutrophils in the glandular lumen or epithelium. Macrophages activated by interferon-γ secrete proinflammatory cytokines and reactive nitrogen species (e.g., nitric oxide). Inducible nitric oxide synthase, which catalyzes the generation of nitric oxide, is overexpressed in macrophages in PIA but not in normal epithelium (Nelson et al, 2003). PIA appears to be a regenerative lesion appearing as a consequence of infection or cell trauma resulting from oxidant damage, hypoxia, infection, or autoimmunity, and its hyperproliferative state may lead to cancer. PIA is often found adjacent to high-grade prostatic intraepithelial neoplasia (HGPIN) or early cancer (Putzi and De Marzo, 2000), and there is an identifiable genetic pathway between PIA, HGPIN, and cancer (Shah et al, 2001; Nakayama et al, 2003; Nelson et al, 2003). The previously described genetic and histologic observations in prostate cancer strongly suggest that compromised cellular defenses against inflammatory oxidants may initiate and/or perpetuate prostatic carcinogenesis. Oxidative stress is mediated by reactive oxygen and nitrogen species that bind DNA and cause mutations, and oxidant stresses from exogenous and endogenous sources are implicated in the accumulation of DNA damage that occurs with aging and subsequently leads to malignant change (Coussens and Werb, 2002). Cellular defense mechanisms against this process include front-line antioxidant enzymes, which scavenge reactive oxygen and nitrogen species and prevent mutations; DNA repair enzymes; and the ability to undergo apoptosis if the DNA damage is too severe to repair. A recent model proposes that inherited and acquired defects in cellular defense mechanisms against infection and oxidative stress allow prostate cancer to develop. This model suggests that, as in other inflammatory-mediated cancers, persistent infection leads to chronic inflammation, and that defects in antioxidant enzymes, DNA repair mechanisms, and apoptosis lead to cancer. Subsequent expression by tumor cells of α-methylacyl-CoA racemase (Kumar-Sinha et al, 2004), an enzyme that oxidizes branched-chain fatty acids from dietary sources, results in generation of hydrogen peroxide, which may contribute to continued oxidative stress, and tumor growth. This model is illustrated in Figure 95–6. In addition to providing a framework for further experimental study, this model gives a theoretical rationale for the use of antioxidants as chemopreventative agents (see later). Like cervical cancer, which is characterized by viral infection coincident with the onset of sexual activity, histologic evidence of preinvasive lesions, and a latency period of 15 to 20 years before the development of invasive cancer, the accumulating histologic, epidemiologic, and genetic evidence suggests the possibility that at least some prostate cancer cases have a viral pathogenesis. The epidemiologic data linking prostate cancer risk to prior infections and the association of a functionally important mutation in the antiviral HPC1/RNaseL gene support this hypothesis. Recently reported studies have identified a previously undescribed γ-retrovirus, xenotropic murine leukemia–related virus (XMRV), from prostate cancer tissue in men with the R462Q allelic variant of HPC1/RNaseL. The initial experiment demonstrated the presence of viral genomes for XMRV in prostate tumors in 8 of 20 patients with the R462Q SNP in RNaseL, but in only 1 of 62 wild-type or heterozygous patients (Urisman et al, 2006). XMRV infections were visualized by immunohistochemistry and fluorescence in-situ hybridizations in prostatic stromal cells adjacent to the tumor. A full-length, replication-competent, and infectious viral molecular clone of XMRV was subsequently cloned from prostate tissue cDNA and has been used for in-vitro studies (Dong et al, 2007a). In addition, XMRV integration sites have been mapped in host chromosomal DNA (Kim et al, 2008), suggesting that modulation of cancer-related gene function by XMRV could be important. The presence of XMRV in prostate tissue has been recently confirmed by two other groups, including evidence of expression of XMRV proteins in malignant prostatic epithelium using a rabbit polyclonal antibody (Fischer et al, 2008; Singh Laboratory, 2010). Together these observations suggest that XMRV infects and persists in the prostate when there is a deficiency in RNaseL activity, providing a proposed mechanism by which XMRV could be oncogenic in genetically susceptible individuals. Other infectious agents have also been implicated in prostate cancer. BK virus (BKV), a member of the polyomavirus family, infects almost 90% of the human population by early childhood and resides in a subclinical persistent state in the urinary tracts of healthy individuals. BKV transforms rodent cells in culture, causes kidney tumors in transgenic mice, and immortalizes primary human cells alone or in the presence of other oncogenes such as c-RAS and adenovirus E1A (Das et al, 2008). The occurrence of mutations in TP53 and RB1 in the early stages of prostate cancer is relatively low, suggesting the possibility that a virus, such as BKV (encoding tumor antigens that inactivate these tumor suppressors), may play a role in the etiology of prostate cancer. BKV DNA has been demonstrated in epithelial cells of benign and PIA ducts of cancerous prostate specimens, and BKV large tumor antigen (Tag) expression has been observed specifically in atrophic epithelial cells, PIA, and prostatic intraepithelial neoplasia (PIN) but not in cancer nor in the epithelium of normal prostates (Das et al, 2004, 2008). These observations suggest a role for BKV in PIA and the early development of prostate cancer. Furthermore, in a study examining the bacterial flora of prostate cancer, Sfanos and Isaacs (2008) demonstrated the presence of 83 distinct microorganisms by sequencing bacterial 16S rDNA in core samples of 30 prostate cancers, most of which were not found by routine culture methods. If XMRV, BKV, or specific bacteria are proven to be oncogenic, their effects may be preventable by development of a protective vaccine. Key Points Inflammation, Infection, and Genetic Susceptibility Gene fusions resulting from chromosomal translocations are the most common genetic alteration in human cancers (Futreal et al, 2004). These were previously thought to be the oncogenic mechanism exclusively limited to hematologic malignancies and sarcomas, as exemplified by the BCR-ABL1 fusion protein in chronic myeloid leukemia. In 2005, recurrent genomic rearrangements in prostate cancer were identified, resulting in the fusion of the 5′ untranslated end of TMPRSS2 (21q22.2, an androgen-responsive, prostate-specific, transmembrane serine protease gene) to members of the ETS family of oncogenic transcription factors (Tomlins et al, 2005). In over two dozen published reports using PSA-screened patient cohorts, the most common fusion identified in localized prostate cancer involves TMPRSS2 fused to ERG (ETS-related gene, 21q22.3) in approximately 50% of patients (Kumar-Sinha et al, 2008). However, this gene fusion may be less common (15%) in population-based cohorts (Demichelis et al, 2007). Gene fusions involving other members of the ETS family, most commonly ETV1 (ETS variant gene 1, 7q21.2), but also ETV4 and ETV5, are likely present in less than 10% of prostate cancer samples collectively (Kumar-Sinha et al, 2008). The TMPRSS2 gene is prostate specific, and is expressed in both benign and malignant prostatic epithelium. TMPRSS2 expression has been shown to be induced by androgens in hormone-responsive prostate cancer cell lines (Lin et al, 1999). Therefore it is hypothesized that TMPRSS2 drives ETS gene overexpression in prostate cancers positive for TMPRSS2-ETS gene fusions (Fig. 95–7) (Kumar-Sinha et al, 2008). Although some studies have demonstrated an association between TMPRSS2-ERG with high pathologic stage (Mehra et al, 2007), higher recurrence rates (Nam et al, 2007), and prostate cancer–specific death (Demichelis et al, 2007), others have failed to report such clinical correlations. It is notable, however, that the presence of TRMPSS-ETS gene fusion was associated with a markedly higher risk of death in men managed by watchful waiting in one Swedish cohort (Demichelis et al, 2007). Currently, the clinical application of this discovery is uncertain. Given that ETS-positive and ETS-negative tumors have distinct morphologic features, expression signatures, and potentially different clinical outcomes, some have proposed classifying prostate cancer as two categories: ETS gene fusion–positive and ETS gene fusion–negative disease (Kumar-Sinha et al, 2008). Because these gene fusions do not occur in benign prostate tissue, a sensitive and specific screening test based on their presence could be developed for the detection of prostate cancer. Eventually, gene fusion status may be used to decide the need for initial or repeat biopsy, for enrollment in therapeutic trials, or for surveillance after therapy. It also follows that targeting ERG activity in fusion-positive prostate cancers may provide an effective therapeutic approach for this subset of patients. Presently, attempts at identifying small molecular inhibitors of the ERG pathway that could serve as therapeutic agents are underway. At this time, the emerging consensus suggests that TRMPSS-related gene fusions are highly specific for the presence of prostate cancer, but their utility as prognostic markers requires further study. Androgens influence the development, maturation, and maintenance of the prostate, affecting both proliferation and differentiation of the luminal epithelium. There is little doubt that exposure of the prostate at key developmental times to androgens plays an important role in prostate carcinogenesis. Androgens are also important in the maintenance of established cancers, as supported by the historical observation that the majority of prostate cancers initially respond to androgen-deprivation therapy, and more recently by results of the Prostate Cancer Prevention Trial, which indicated that inhibition of the conversion of testosterone to the more potent dihydrotestosterone by finasteride reduces the incidence of prostate cancer by approximately 25% (Thompson et al, 2003). In addition, genetic polymorphisms of the androgen receptor (AR) (Balic et al, 2002; Taplin et al, 2003), the 5α-reductase type 2 isoenzyme (Makridakis and Reichardt, 2004), and the genes involved in biosynthesis of testosterone have also been implicated in prostate carcinogenesis (Chang et al, 2002). High serum androgen levels have long been hypothesized to be a risk factor for prostate cancer. However, studies examining this association have been inconsistent, with some studies finding an association between specific hormones and prostate cancer risk. Recently, a collaborative group performed a pooled analysis of 18 prospective studies assessing this association using data from 3886 men with prostate cancer and 6438 control subjects (Endogenous Hormones, Prostate Cancer Collaborative Group, 2008). The collaborative analysis failed to detect an association between the risk of prostate cancer and serum concentrations of testosterone, calculated free testosterone, dihydrotestosterone, dehydroepiandrostenedione sulfate, androstenedione, androstanediol glucuronide, estradiol, or calculated free estradiol. The only positive finding in this study was a modest inverse association between prostate cancer risk and serum concentration of sex-hormone binding globulin. This study suggests that one-time measurements of serum sex hormones in adulthood are not a good measure of prostate cancer risk. Estrogens have both direct and indirect effects on prostatic growth and development and have been postulated to play a role in prostate cancer initiation and progression. Traditionally, estrogens have been considered protective against prostate cancer and have been used as a treatment for advanced disease. This treatment effect is primarily through a negative feedback on the hypothalamo-pituitary-gonadal axis, and also through a direct inhibitory effect of estrogens on prostate epithelial cell growth. However, there is increasing evidence from animal studies that estrogens may act as procarcinogens in the prostate (Leav et al, 1988). Aromatase-knockout mice cannot produce 17β-estradiol locally in the prostate, and despite elevated testosterone and dihydrotestosterone, they do not develop prostate cancer (McPherson et al, 2001). There is evidence across multiple species that estrogens acting through stromal estrogen receptor (ER) α may contribute to prostate carcinogenesis and cancer progression (Prins and Korach, 2008). ERα expression is silenced in early prostate cancers, and re-emerges with disease progression. Prostate epithelial ERβ may play an important role in initiation of prostate cancer, with loss of ERβ potentially contributing to disease progression in organ-confined disease (Prins and Korach, 2008). In addition, the re-emergence of ERβ expression in metastatic prostate cancer also suggests a potential role in progression to castrate-resistant disease. Age-related prostatic disease parallels increases in serum estrogen levels, and there is a low incidence of prostate cancer in cultures with diets rich in phytoestrogens (Denis et al, 1999). However, the data on serum estrogen levels and prostate cancer risk are mixed (Barrett-Connor et al, 1990; Gann et al, 1996; Chen et al, 2003). Complicating interpretation of serum measurements is the fact that estradiol can be produced from testosterone by intraprostatic aromatase (Risbridger et al, 2003). Insulin-like growth factor (IGF)-1 is a peptide hormone that promotes growth in adolescence and childhood and is correlated with adult lean body mass (Severson et al, 1988). IGF-1 promotes proliferation and inhibits apoptosis in normal prostate and tumor cells in vitro (Cohen et al, 1991; Cohen et al, 1994). IGF-1 circulates bound to binding proteins, the most prevalent of which is insulin-like growth factor binding protein (IGFBP)-3. In the prostate IGFBP-3 promotes apoptosis and may mediate growth inhibition by 1,25-dihydroxyvitamin D (Boyle et al, 2001). IGFBP-3 can be cleaved by PSA, reducing its pro-apoptotic activity (Koistinen et al, 2002). Results of epidemiologic studies suggest an association between IGFs and their associated binding proteins (IGFBPs) with prostate cancer risk. A recent combined analysis of individual patient data from 12 studies attempted to quantify the magnitude of the association (Roddam et al, 2008). The study included data on 3700 men with prostate cancer and 5200 control subjects. They found a positive correlation between serum concentration of IGF-1 and subsequent prostate cancer risk. The OR in the highest versus lowest quintile was 1.38 (95% CI = 1.19 to 1.60). Serum IGFBP-3 concentration was not found to be independently associated with prostate cancer risk; the observed association was secondary to its association with IGF-1 levels. Neither IGF-2 nor IGFBP-2 concentrations were associated with prostate cancer risk. Leptin, a peptide hormone produced by adipocytes, contributes to the control of body weight by appetite suppression and modulating energy use (Friedman, 2002). Obese men become leptin resistant and exhibit elevated plasma leptin (Chu et al, 2001). Epidemiologic studies assessing the association between circulating leptin concentrations with prostate cancer have yielded mixed results (Chung and Leibel, 2006). A genetic study found that prostate cancer patients were almost 5 times more likely to carry a polymorphism that results in increased leptin expression compared with controls (Ribeiro et al, 2004). There is significant evidence, however, suggesting that leptin plays a role in the development of advanced prostate cancer (Ribeiro et al, 2006). Leptin has been shown to stimulate proliferation of the androgen-independent prostate cancer cell lines DU145 and PC-3 (Somasundar et al, 2004; Deo et al, 2008). In addition, leptin appears to induce expression of vascular endothelial growth factor (VEGF) and basal fibroblast growth factor (BFGF), and stimulate cell migration (Frankenberry et al, 2004). Energy imbalance, perhaps mediated by leptin, is emerging as a possible contributor to the progression of prostate cancer to metastasis and death (Calle et al, 2003; Platz et al, 2003). Vitamin D (1,25 dihydroxyvitamin D3) is an essential vitamin that is a part of the steroid hormone superfamily. Human sources include both dietary intake and conversion from an inactive to active vitamin D in the skin through sunlight exposure. Interest in vitamin D as a determinant of prostate cancer risk comes from several epidemiologic observations (Peehl et al, 2003): In addition, prostate cancer cells express the vitamin D receptor, and several studies have demonstrated an antiproliferative effect of vitamin D on prostate cancer cell lines by inducing cell cycle arrest (Krishnan et al, 2003). Many studies show no or a weak association between vitamin D levels and prostate cancer risk (Chan and Giovannucci, 2001; Freedman et al, 2002; Jacobs et al, 2004; Ahn J et al, 2008). The Cancer Prevention Study II Nutrition Cohort, a prospective cohort of 65,321 men demonstrated a modestly increased relative risk of 1.2 for total calcium intake (dietary and with supplements) and 1.6 for high dietary calcium intake alone (≥2000 vs. <700 mg/day), but not for dairy intake (Rodriguez et al, 2003). The results suggest that very high calcium intake, above daily recommendation, may modestly increase risk. These conflicting results regarding vitamin D, calcium, and prostate cancer risk may be explained by variants in the vitamin D receptor (VDR). Polymorphisms resulting in a VDR with lower activity have been associated with increased risk for prostate cancer, as well as with increased risk of biochemical recurrence following radical prostatectomy (Oakley-Girvan et al, 2004; Williams et al, 2004; John et al, 2005). Sexual activity has been hypothesized to expose the prostate to infectious agents, which may increase the risk of prostate cancer, akin to the causal relationship between HPV and cervical cancer in women. Some studies have found a link between sexually transmissible infections (STIs) and prostate cancer risk (Fernandez et al, 2005; Sarma et al, 2006), although not consistently (Patel et al, 2005; Huang et al, 2008). A meta-analysis of 6022 cases of prostate cancer and 7,320 controls found significant associations between prostate cancer and any STI (odds ratio [OR] = 1.48, 95% CI = 1.26 to 1.73), gonorrhea (OR = 1.35, 95% CI = 1.05 to 1.83), and human papillomavirus (OR = 1.39, 95% CI = 1.12 to 2.06) but not for syphilis (Taylor et al, 2005). Studies have also suggested a protective association between prostate cancer and frequency of ejaculation, with RR ranging from 0.66 to 0.89 (Giles et al, 2003; Leitzmann et al, 2004). In the Giles study, the protective effect was seen in men in their 20s, who reported greater than 5 ejaculations per week; the large prospective cohort study by Leitzmann demonstrated a protective effect for men in their 20s and 40s, reporting greater than or equal to 21 ejaculations per month in the previous year and as a lifetime average. The biologic basis for this effect is not known. A relationship between vasectomy and prostate cancer risk was initially suggested in 1993 with a relative risk of 1.6 based on two large cohort studies (Giovannucci et al, 1993a, 1993b). Risk increased with time so that men who underwent vasectomy at an early age had a higher risk. A recent meta-analysis reported a pooled relative risk of 1.37, with a linear trend suggesting a 10% increase for each additional 10 years since vasectomy (Dennis et al, 2002a). On the other hand a large, population-based, case-control study found no association between prostate cancer and vasectomy, nor with time since vasectomy (Cox et al, 2002
Epidemiology
Incidence and Mortality Trends
Incidence
INCIDENCE*
MORTALITY*
White
161.4
25.6
African-American
255.5
62.3
Hispanic/Latino
140.8
21.2
Asian-American and Pacific Islander
96.5
11.3
American Indian and Alaska Native
68.2
21.5

Mortality
Racial Differences
Worldwide Incidence and Mortality
Age at Diagnosis
Stage at Diagnosis
Effect of Screening on Mortality
Risk Factors
Familial and Genetic Influences
FAMILY HISTORY
RELATIVE RISK
95% CONFIDENCE INTERVAL
None
1
Father affected
2.17
1.90-2.49
Brother affected
3.37
2.97-3.83
First-degree family member affected, age less than 65 years at diagnosis
3.34
2.64-4.23
Greater than two first-degree relatives affected
5.08
3.31-7.79
Second-degree relative affected
1.68
1.07-2.64
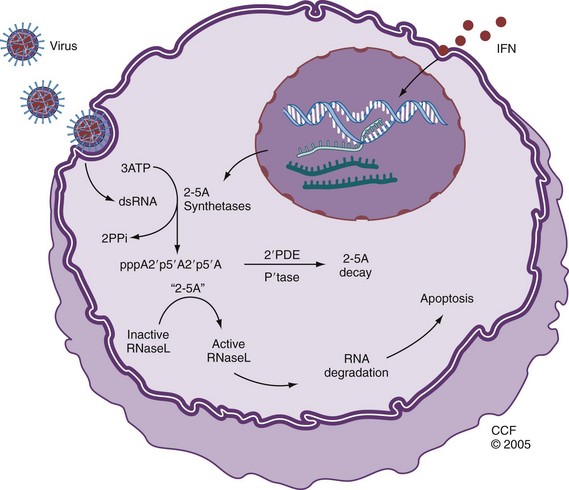
Inflammation, Infection, and Genetic Susceptibility
Molecular Epidemiology
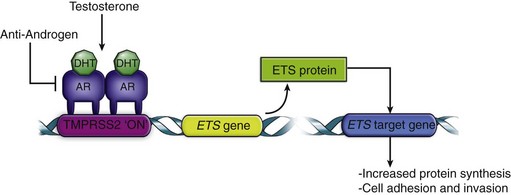
Gene Fusions
Androgens
Estrogens
Insulin-Like Growth Factor Axis
Leptin
Vitamin D, Vitamin D Receptor, and Calcium
Other Influences
Sexual Activity/Sexually Transmitted Diseases
Vasectomy
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
Surgical Procedures for Sphincteric Incontinence in the Male: The Artificial Genitourinary Sphincter and Perineal Sling Procedures
 Prosthetic Surgery for Erectile Dysfunction
Prosthetic Surgery for Erectile Dysfunction
 Neuropathic Dysfunction of the Lower Urinary Tract
Neuropathic Dysfunction of the Lower Urinary Tract
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Epidemiology, Etiology, and Prevention of Prostate Cancer
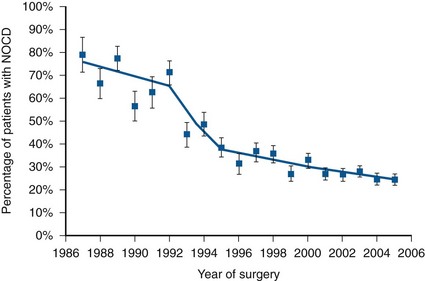
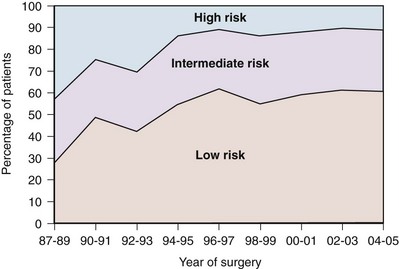






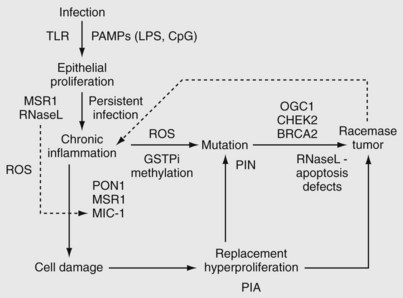
• Chronic inflammation leading to cellular hyperproliferation to replace damaged tissue contributes to the development of infection-associated cancers, possibly including prostate cancer.
1. Men living in northern latitudes with less exposure to sunlight-derived ultraviolet exposure have a higher mortality rate from prostate cancer.
