Neonate
Infant
Child
I
Congenital transport and enzymatic deficiencies
(A) Carbohydrates
Glucose–galactose malabsorption
Disaccharidase deficiency
Trehalase deficiency
Congenital lactase deficiency
Sucrase–isomaltase deficiency
Fructose malabsorption
(B) Lipids
Abetalipoproteinemia/hypobetalipoproteinemia
Chylomicron retention disease
Wolman’s disease
(C) Amino acids
Lysinuric protein intolerance
(D) Electrolytes and trace elements
Congenital chloride diarrhea
Acrodermatitis enteropathica (zinc)
Congenital sodium diarrhea
Menkes disease (copper)
(E) Vitamins
Congenital folate malabsorption
Disorders of B12 transport (CblC deficiency)
(F) Bile acids
Primary bile acid Malabsorption
II
Primary epithelial abnormalities
Microvillus inclusion disease
Enterocyte heparan sulfate deficiency
“Tufting” enteropathy
Enteroendocrine dysgenesis
III
Inflammatory and immune-mediated disorders
Autoimmune enteropathy
Chronic inflammatory bowel disease
Cow’s milk and soy protein intolerance
Eosinophilic enteropathy
Enterocolitis with phenotypic and immune abnormalities
Gluten-sensitive enteropathy
Agammaglobulinemia
Other immunodeficiency syndromes
Severe combined immunodeficiency
IV
Infections
Post-infectious enteritis
Bacterial overgrowth/blind loop
Clostridium difficile
Amebiasis
Giardia lamblia infestation
Cryptosporidium
HIV infection
V
Anatomical disorders
Necrotizing enterocolitis
Malrotation
Lymphangiectasia
VI
Systemic disorders and tumors
Infantile systemic hyalinosis
Myofibromatosis
Langerhans cell histiocytosis
Glycogen storage disorder type Ib
Erythrophagocytic lymphohistiocytosis
Disorders of glycosylation
Lymphoproliferative disorders
Functional tumors (neuroblastoma)
VII
Motility disorders
Aganglionosis and variants
Chronic pseudo-obstruction
VIII
Pancreatic disorders
Shwachman’s disease
Cystic fibrosis
4.1.1 Mechanisms of Diarrhea
Diarrhea results from incomplete absorption of water from the gut lumen and may be due to impaired electrolyte absorption resulting from mucosal damage, excessive secretion of electrolytes (from abnormalities of enterocyte electrolyte transporters), or from osmotic retention of water. It has been estimated that approximately 95 % of the fluids and secretions which pass the ligament of Treitz per day is reabsorbed by the large and small bowel, resulting in a stool output of about 5–10 g/kg/day. The control of water movement across the gut is the result of a delicate balance in the bidirectional flow of fluid across the enterocyte membrane and is intimately linked with solute transport. A complex series of energy-dependent pumps, under the regulatory influences of the enteric nervous system, combine to drive Na+ and nutrients into the enterocyte, with passive absorption of water. Water is cotransported with glucose or amino acids or enters via acid–base exchangers such as the Na+/H+ exchanger located on the brush border of enterocytes. Absorption of water mediated by the Na+/glucose cotransporter (SGLT1) results in the absorption of two molecules of sodium and 225 molecules of water for each molecule of sugar absorbed (Field 2003). The apically located Na+/H+ exchangers are called NHE1-3 and are under the control of the NHE genes. These transporters act via cGMP and Ca2+, such that increased intracellular Ca2+ inhibits the exchanger, reduces Na+ absorption, and increases Cl− secretion (Surawicz 2010). Chloride and potassium channels permit egress of their respective ions into the extracellular space, to be either absorbed into the vasculature or secreted into the gut lumen. The cystic fibrosis transmembrane conductance regulator (CFTR) is one such important regulator of chloride transport across the cell. Its absence or defect, as in cystic fibrosis, results in a loss of Cl– secretion and consequent dehydration of the mucus. Substances synthesized by the intestines such as the guanylin-family hormones and enterotoxins produced by bacterial pathogens such as enterotoxigenic E. coli bind to guanylate cyclase C (GC-C) receptors located on the surface of the enterocytes. Binding to these receptors causes increased intracellular levels of cyclic guanosine monophosphate (cGMP), eliciting a signaling cascade which results in increased intracellular Ca++ levels, decreased Na+ reabsorption, and increased chloride secretion through the CFTR channel (Surawicz 2010; Fiskerstrand et al. 2012). Mutations in the gene coding for the guanylate cyclase C receptor, GUCY2C, have recently been linked to cases of familial diarrhea (Fiskerstrand et al. 2012). The enteric nervous system is also known to play a role in intestinal absorption and secretion. Enteroendocrine (EC) cells, activated by nutrients, changes in pH, or solute concentrations, are believed to transmit information about luminal contents to sensory neurons via their terminal endings in the intestinal wall. EC cells also release mediators such as serotonin to intrinsic primary afferent neurons (IPANs) in the submucosal and myenteric plexi. These secrete acetylcholine, which affects peristalsis and also results in dilatation of blood vessels with increased blood flow and release of water, NaCl, and mucus from the intestinal glands into the lumen (Surawicz 2010).
Secretory diarrhea results from abnormal secretion of electrolytes into the lumen. It can result from congenital defects of ion absorption such as Na+/Cl− diarrhea, from loss of intestinal surface area due to diffuse mucosal disease, and from abnormal circulating mediators, including enteric hormones, neuropeptides, and bacterial enterotoxins. Secretory diarrhea does not resolve with bowel rest. On the other hand, osmotic diarrhea results from the presence of unabsorbed or poorly absorbed solute in the gut. There is an osmotic “gap” in stool analysis (2 × [Na+ + K+] < 280 mOsm) signifying the presence of unmeasured particles, usually sugars and low molecular weight compounds. Typically, the diarrhea resolves with bowel rest or removal of the offending solute.
Motility disorders can cause diarrhea either when enhanced, as a result of abnormal mediators, or when decreased, by allowing bacterial overgrowth resulting in excessive deconjugation of bile acids. In the most severe cases diarrhea may even be due to overflow (Fine 1998). Malabsorption is caused by a wide range of entities that is frequently associated with chronic diarrhea and failure to thrive and results from defective mucosal absorption of dietary constituents. It is different from maldigestion, which is the abnormal hydrolysis of nutrients, but in common clinical practice, malabsorption encompasses both. Malabsorption of proteins, carbohydrates, and fat can occur alone or in combination, depending on the etiology. Sprue is an older term which is essentially synonymous with malabsorption. Protein-losing enteropathy results from excessive loss of protein into the gastrointestinal tract, resulting in hypoalbuminemia and peripheral edema. Lymphangiectasia, either primary or secondary, is an important cause of protein-losing enteropathy.
4.1.2 The Intestinal Intercellular Junction
The intestinal epithelium functions both as a barrier to foreign antigens and microorganisms as well as a selective filter allowing access of nutrients, electrolytes, and water to the internal milieu. This selective permeability is mediated by two major routes: the transcellular and paracellular pathways. The transcellular pathway is mediated by solute-specific transporters allowing passage of the solute through the cell, and the paracellular pathway is mediated by the intercellular junctional complex. The intercellular junction is currently the focus of great interest in intestinal physiology and disease; an increasing number of experimental and natural diseases causing diarrhea have been correlated with abnormalities of one or more of the molecular junctional components. Several excellent recent reviews are available (Assimakopoulos et al. 2011; Marchiando et al. 2010; Groschwitz and Hogan 2009). In brief, the junctional complex consists of three major components: tight junctions (TJs), the subjacent adherens junction, or zonula adherens, and the desmosomes (Fig. 4.1). The most basal is the desmosome, consisting of a closely woven set of tonofilaments anchored to an electron-dense plaque, acting mainly as an adhesion point between the cells. The most apically situated is the tight junction or zonula occludens, which seals the paracellular space, whereas the adherens junction and desmosomes provide the bonds necessary to maintain cellular proximity and allow the assembly of the tight junction. More than 50 different tight junction-associated proteins have been described, which fall into four broad classes: the transmembranous occludin and claudins, the latter consisting of a family of over 20 proteins; the junctional adhesion molecule; and tricellulin (Assimakopoulos et al. 2011). These interact with cytoplasmic plaques that consist of cytosolic proteins including the zonula-occludens proteins ZO-1, ZO-2, and ZO-3, which function as adaptors between the tight junction proteins and myosin and actin filaments within the enterocyte. Diseases associated with mutations in tight junction proteins have been described. The syndrome of neonatal sclerosing cholangitis and ichthyosis has been associated with claudin-1 gene (CLDN1) mutations (Hadj-Rabia et al. 2004), whereas mutations in the gene coding for claudin-16 (CLDN16) cause hypomagnesemia with hypercalciuria and visual impairment (Marchiando et al. 2010). A number of pathogenic bacterial strains such as Vibrio cholerae, Shigella, Clostridium perfringens and difficile, as well as Enteropathogenic E. coli are known to secrete toxins which disrupt the intestinal tight junction (Assimakopoulos et al. 2011). Vibrio cholerae expresses a zonula occludens toxin, resulting in disassembly of tight junctions (Wang et al. 2000). A reduction in the expression of tight junction proteins such as occludin, claudin-4, and claudin-5 has been observed in human Norovirus and other enteric viral infections (Troeger et al. 2009). Occludin has been reported to be decreased in the actively inflamed mucosa of Crohn’s disease and ulcerative colitis (Gassler et al. 2001). The purported target of the Vibrio cholerae toxin, zonulin, has been proposed as an initiating factor in the pathogenesis of celiac disease (Wang et al. 2000). In vitro studies have demonstrated that treatment of Caco-2 cells with gliadin downregulates the expression of occludin, claudin-3 and claudin-4, E-cadherin, and ZO-1 (Cereijido et al. 2007). Recent evidence also suggests that allergens may disrupt the tight junction. A cysteine protease from fecal pellets of house dust mites has been reported to cleave occludin and claudin-1 and induce the reorganization of ZO-1 (Cereijido et al. 2007).
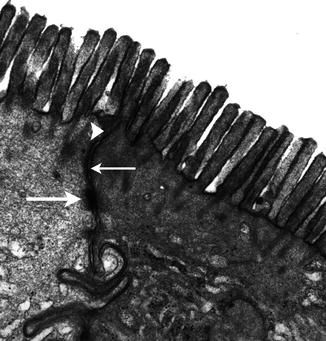
Fig. 4.1
The intercellular junctional complex of the enterocyte. The arrowhead indicates the tight junction (TJ), the thin arrow the adherens junction, or zonula adherens, and the thick lower arrow the desmosomes. Uranyl-lead, X 22,000
Located between the desmosomes and the zonula occludens is the zonula adherens (ZA), principally composed of the cadherin–catenin–actin complex. E-cadherin is a transmembrane protein which is part of the cadherin superfamily and the predominant intercellular adhesion molecule expressed by epithelial cells. E-cadherin binds to either β- or γ-catenin, which in turn is linked to the actin microfilament system via α-catenin. Transgenic mice deficient in E-cadherin have a severe colitis with increased intestinal permeability (Jawhari et al. 1997). E-Cadherin and β-catenin are reportedly decreased in celiac disease (Barshack et al. 2001). Furthermore, β-catenin, via its interaction with the product of the APC gene, is an important mediator of epithelial activation and may play a pivotal role in intestinal dysplasia and tumorigenesis (Venesio et al. 2003). Disorders of the desmosomal complex have been primarily implicated in dermatological diseases; however, some appear to also involve the gut. Epidermolysis bullosa associated with pyloric atresia (EB-PA) is due to mutations in the ITGA6 and ITGB4 genes which code for the corresponding units of α6β4 integrin, an adhesion molecule expressed in a variety of epithelia, including the epidermis and GI tract, where it forms an integral part of the hemidesmosomes (Chung and Uitto 2010). Abnormalities of desmosomes have also been described in “tufting” enteropathy (Goulet et al. 2007).
4.2 The Intestinal Biopsy: General Principles
4.2.1 Biopsy Sampling and Indications
As pediatric esophagogastroduodenoscopy (EGD) has evolved into a routine outpatient procedure over the last few decades, indications for its use have correspondingly changed. At the Children’s Hospital of Philadelphia, the first-time EGD rate increased 12-fold in a 20-year interval between 1985 and 2005, with isolated abdominal pain replacing GI bleeding as the most frequent indication (Franciosi et al. 2010). As one would expect, the proportion of biopsies with histologic abnormalities also correspondingly declined (Franciosi et al. 2010). In children less than 1 year of age, the most frequent indications for endoscopy were diarrhea, failure to thrive, reflux, and rectal bleeding, according to one recent study (Volonaki et al. 2011). Histologic abnormalities were detected in two-thirds of cases. In current gastrointestinal practice, endoscopic forceps biopsies have replaced the Crosby suction biopsies because of greater ease of the procedure, patient comfort, avoidance of radiation exposure, direct visualization of the GI tract, which allows more precise targeting of the mucosal lesion, and the possibility of obtaining multiple biopsies from several sites. Numerous studies have shown that the histologic quality and interpretation of endoscopic forceps biopsies and capsule biopsies are equivalent (Achkar et al. 1986; Dandalides et al. 1989; Granot et al. 1993; Mee et al. 1985; Thomson et al. 1999). In the study by Volonaki et al. mentioned previously, only 2 % of biopsies in children less than 1 year of age were found to be insufficient for diagnosis (Volonaki et al. 2011). In addition to obtaining biopsies for routine histology, samples may also be snap-frozen (for disaccharidase analysis) or submitted for electron microscopy (for confirmation of microvillus inclusion disease). Biopsies from both proximal and distal duodenum are recommended, including biopsies of endoscopically normal mucosa, as many disorders affecting the duodenum have a focal distribution. In children, for example, the lesions in gluten-sensitive enteropathy (GSE) may be patchy, and villus atrophy may coexist with normal mucosa (Bonamico et al. 2004). Furthermore, villus atrophy may be limited to or most severe in the duodenal bulb at the time of diagnosis (Bonamico et al. 2004; Levinson-Castiel et al. 2011). Sampling endoscopically normal-appearing mucosa is recommended, as it may help assess the “background” features of the mucosa, and histologic examination may reveal clinically relevant findings unsuspected by the endoscopist (i.e., granulomas) (De Matos et al. 2008; Kori et al. 2003). A recent pediatric study found that routine duodenal biopsies performed for indications such as gastroesophageal reflux, vomiting, abdominal pain, anemia and during the evaluation of Crohn’s disease yielded clinically unsuspected pathologic findings in about 17 % of cases; conversely, a normal-appearing mucosa did not rule out abnormal histology (Kori et al. 2003).
4.2.2 Interpretation of the Duodenal Biopsy
Villi appear in the duodenum during the 8th week and crypts of Lieberkuhn are noted during the 9th week (Fig. 4.2), spreading caudally and arriving in the distal ileum by the 14th week. The definitive histologic features of the duodenum are established by the 14th week post-fertilization, and its histology closely resembles that of the newborn by 20 weeks (Fig. 4.3). The transition from pyloric to duodenal mucosa is gradual in some cases, and duodenal-like villi may be found in the distal pylorus and pyloric-like epithelium may be found in the duodenum. An adequate biopsy should have four to five well-oriented villi in a row, and observation of four normal villi in a row is generally an indication that the specimen is normal (Gramlich and Petras 2007) (Fig. 4.4). The normal villus height–crypt depth ratio in the duodenum is about 3:1 to 5:1. It is somewhat greater in adults (Whitehead 1997) and higher in the distal as compared to the proximal duodenum (Korn and Foroozan 1974) (Fig. 4.5). Whether genetically determined or resulting from environmental factors such as endemic infections, “normal” histologic findings in the small bowel also vary according to geography and race. Somewhat shorter villi and increased lamina propria mononuclear cells are reported in healthy individuals from developing countries as compared to industrialized nations (Bennett et al. 1985; Cook et al. 1969; Lindenbaum et al. 1971). The villus epithelium consists of absorptive enterocytes, characterized by basally located nuclei and a smooth brush border demonstrable by the periodic acid–Schiff (PAS) stain, as well as by immunohistochemical markers such as CD10. Goblet cells are interspersed between the enterocytes and contain an Alcian-blue-positive sialomucin (Fig. 4.6). Paneth cells are located at the base of the intestinal crypts and are easily recognized by the presence of eosinophilic cytoplasmic granules. Paneth cells form part of the innate immune system of the bowel, secreting substances such as defensins, lysozyme, and tumor necrosis factor-α (TNF-α), which play a role in protecting the gut from pathogenic microorganisms. Enteroendocrine cells are best demonstrated by immunohistochemical stains which label nonspecific neurosecretory granules such as chromogranin or specific neurosecretory products. These latter three subsets of mucosal cells appear to differentiate prior to 14 weeks under the control of transcription factors such as Math1 (Yang et al. 2001; Shroyer et al. 2007). The upper limit of intraepithelial lymphocytes (IELs) in the duodenum is estimated to be 20/100 epithelial cells by H+E staining, in contrast to the normal upper limit of 40 per 100 epithelial cells established 30 years ago mainly on the basis of studies of the jejunum (Ferguson and Murray 1971). Using immunostaining for CD3 to identify IELs, the upper limit of normal is reported as 25/100 epithelial cells in biopsies from the distal duodenum, with 26–29 CD3+ IELs considered borderline and 30 or more CD3+ IELs considered pathological (Veress et al. 2004) (Fig. 4.7). These CD3+ IELs are CD8+ T lymphocytes which belong to the MALT (mucosa-associated lymphoid tissue) system. The vast majority normally express the αβ T-cell surface receptor (TCR), and a small minority (<10 %) the γδ receptor (Kutlu et al. 1993). TCRγδ-expressing IELs are believed to mediate induction of oral tolerance. The lamina propria contains a mixture of plasma cells (mostly IgA secreting), CD4+ T lymphocytes, and lesser numbers of eosinophils, histiocytes, and mast cells. Newborns usually lack plasma cells in the first week of life, gradually acquiring them during the first month of life, with IgM-containing plasma cells predominating. By 3 months of life, IgA plasma cells predominate. Normal numbers of plasma cells and ratios of IgA/IgM/IgG are attained by the first year of life (Perkkio and Savilahti 1980).
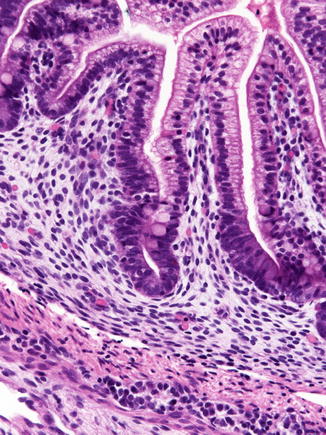
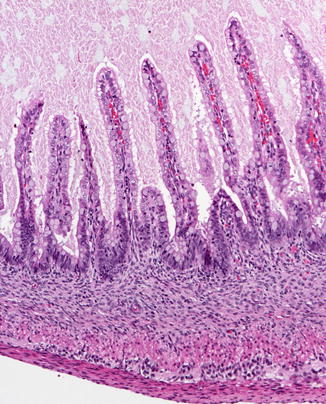
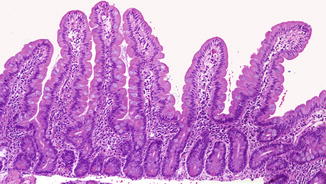
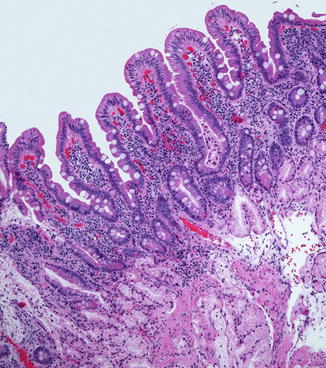
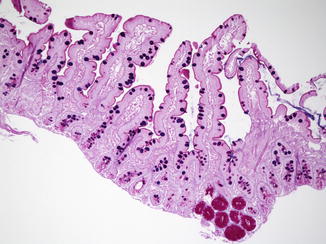
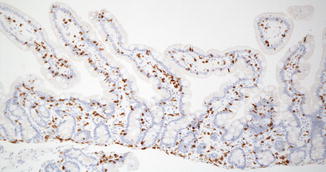
Fig. 4.2
The duodenum at 12 weeks post-fertilization. The epithelium is villiform throughout the gastrointestinal tract. Goblet cells have appeared. Crypts of Lieberkuhn are becoming recognizable. The primordial epithelium seen previously is differentiating into the crypts and surface epithelium. Brunner’s glands have not yet differentiated (With kind permission from Springer Science + Business Media: Ernst et al. (2011))
Fig. 4.3
Small intestine at 19 weeks. The histology of the mucosa is nearly identical to that of a full term infant. The mouths of the crypts of Lieberkuhn are well illustrated. The muscularis mucosae is not present. The muscularis propria and Auerbach’s plexus are well developed (With kind permission from Springer Science + Business Media: Ernst et al. (2011).)
Fig. 4.4
Normal well-oriented duodenal biopsy in a 3-month-old child with at least four normal villi in a row and a villus/crypt ratio of 3–4:1 (H+E, ×100)
Fig. 4.5
Normal duodenal bulb. Note the presence of Brunner’s glands in the mucosa. The villi appear shorter and somewhat blunter than in the more distal duodenum; compare with Fig. 4.4 (H+E, ×100)
Fig. 4.6
Normal duodenum. Intestinal goblet cells are stained purple, whereas the brush border and the Brunner’s glands stain magenta {alcian blue–periodic acid–Schiff (PAS), ×100}
Fig. 4.7
Immunostain for CD3 in a normal duodenum highlights the lymphocytes in the lamina propria and contrasts well with epithelial cells and other stromal elements (avidin–biotin immunoperoxidase, ×100)
Histologic evaluation of the small bowel biopsy should proceed systematically, to include (1) adequacy of the sampling, as determined by the number and site of the biopsy specimens; (2) assessment of the villus architecture with evaluation of the villus to crypt (V:C) ratio; (3) evaluation of the crypts (hyperplasia, hypoplasia); (4) the appearance of the surface enterocytes and their brush border; (5) intraepithelial lymphocytes; (6) the cellularity of the lamina propria (plasma cells may be absent or decreased in some primary immunodeficiencies); and (7) the presence of microorganisms (Giardia, cryptosporidia). Accurate evaluation of villus morphology requires careful attention to proper orientation of the specimen. Some laboratories have found that orientation of the specimen on filter paper prior to fixation is helpful (Serra and Jani 2006). We have found that rapid immersion of the specimen in fixative is preferable to having the endoscopist place the biopsy on filter paper, which may result in excessive stretching or drying of the specimen. Poor quality of the specimen, procedural artifacts, and tangential sectioning may lead to misinterpretations (Table 4.2) (Fig. 4.8).
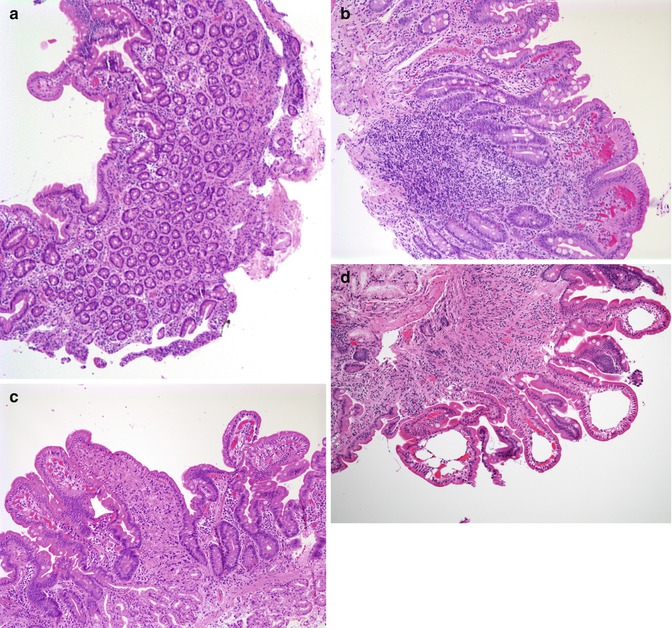
Table 4.2
Pitfalls in the evaluation of endoscopic intestinal biopsies
Event | Feature |
|---|---|
Poorly oriented section | Apparent lack of villus structures |
Preparation-related changes | Clumping of inflammatory cells, epithelial degenerative changes, focal neutrophilic infiltrates, superficial hemorrhages |
Insufflation of air at endoscopy | Air spaces in mucosa (pseudolipomatosis) |
Suction trauma | Subepithelial blebs and dilated lymphatic spaces |
Squeeze and crush artifacts | Crushed Brunner’s glands mistaken for pathologic condition |
Inadequate sampling depth | Absence of submucosa |
Lymphoid aggregate | Distortion of villus architecture |
Fig. 4.8
Artifacts in duodenal biopsy specimens. (a) Tangential sectioning of a duodenal biopsy can give a false impression of villus atrophy and flattening. A tangential section can be identified by the elliptical or circular appearance of the crypts cut in cross section. (b) Broadening and blunting of the villi over a hyperplastic lymphoid nodule are noted in this duodenal biopsy from a 4-year-old patient. (c) Squeezed Brunner’s glands may result in a false impression of a histiocytic infiltrate. (d) “Pneumatosis” resulting from insufflation of air into the mucosa during endoscopy. These spaces may mimic dilated lacteals, from which they can be distinguished by the lack of an endothelial lining and absence of immunoreactivity with D2-40 (all H+E, ×100)
Intestinal biopsy findings in some entities, such as congenital transport disorders, are associated with a normal biopsy, whereas others, such as autoimmune enteropathy or celiac disease, have variable degrees of villus atrophy with or without inflammation. Some diseases, such as abetalipoproteinemia or microvillus inclusion disease, present characteristic findings on an intestinal biopsy (Table 4.3).
Table 4.3
Diagnostic findings on small intestinal biopsies
Intestinal biopsy | Differential diagnosis |
|---|---|
Normal | Sucrase–isomaltase deficiency |
Congenital lactase deficiency | |
Fructose malabsorption | |
Glucose–galactose malabsorption | |
Congenital Na+/CI– diarrhea | |
Nonspecific inflammatory lesions | |
Villus atrophy | Celiac disease |
Autoimmune enteropathy | |
Variable villus atrophy | Protracted infectious diarrhea |
Immunodeficiency states (most) | |
Bacterial overgrowth | |
Cow’s milk or soy protein intolerance | |
Chronic inflammatory bowel diseases | |
Specific lesions | |
Fat-filled enterocytes | Abetalipoproteinemia |
Anderson’s disease | |
Ectatic lymphatics | Lymphangiectasia |
Dense inspissated mucus | Cystic fibrosis |
Epithelial abnormalities | Microvillus inclusion disease |
Tufting enteropathy | |
Enterocolitis cystica | |
Eosinophils | Eosinophilic gastroenteritides |
Absence or paucity of inflammatory cells | Severe combined immunodeficiency |
Agammaglobulinemia | |
Infectious agent | Giardia lamblia infection |
Cryptosporidiosis | |
4.3 Congenital Disorders of Intestinal Digestion, Absorption, and Transport
Specific gene defects associated with various disorders of substrate transport have been recently characterized (Table 4.4). These are instrumental in providing a picture of enterocyte function at the molecular level. Except for the disorders associated with fat processing, small intestinal biopsies in these cases are generally normal or only very slightly abnormal. A normal-appearing small bowel mucosa from a patient with prolonged diarrhea, especially a young infant, should alert the clinician to a consideration of these entities. Only those disorders with significant histologic findings or with confounding clinical features will be discussed in this Section.
Table 4.4
Molecular basis of disorders of digestion, absorption, and transport
Disease | Gene | Location | Function |
|---|---|---|---|
Disaccharidase deficiency | |||
Congenital lactase deficiency | LCT | 2q21 | Lactase–phlorizin hydrolase activity |
Sucrase–isomaltase deficiency | EC 3.2.1.48 | 3q25-q26 | Isomaltase–sucrase |
Maltase–glucoamylase deficiency | MGAM | 7q34 | Maltase–glucoamylase activity |
Ion and nutrient transport defects | |||
Glucose–galactose malabsorption | SGLT1 | 22q13.1 | Na+/glucose cotransporter |
Fructose malabsorption | GLUT5 | 1p36 | Fructose transporter |
Fanconi–Bickel syndrome | GLUT2 | 3q26 | Basolateral glucose transporter |
Cystic fibrosis | CFTR | 7q31.2 | cAMP-dependent CL- channel |
Acrodermatitis enteropathica | SLC39A4 | 8q24.3 | Zn2+ transporter |
Congenital chloride diarrhea | DRA | 7q22-q31.1 | CL-/base exchanger |
Congenital sodium diarrhea | SPINT2* | 19q13.1 | Serine-protease inhibitor |
Lysinuric protein intolerance | SLC7A7 | 14q11 | Hydrolyzes endo-/exopeptidases amino acid basolateral transport |
Congenital bile acid diarrhea | ABAT | 13q3 | Ileal Na+/bile salt transporter |
Pancreatic insufficiency | |||
Enterokinase deficiency | PRSS7 | 21q21 | Proenterokinase |
Trypsinogen deficiency | PRSS1 | 7q35 | Trypsinogen synthesis |
Pancreatic lipase deficiency | PNLIP | 10q26.1 | Hydrolyzes triglycerides to fatty acids |
Lipid trafficking | |||
Abetalipoproteinemia | MTP | 4q22 | Transfer lipids to apolipoprotein |
Hypobetalipoproteinemia | APOB | 2p24 | Apolipoprotein that forms chylomicrons |
Chylomicron retention disease | SAR1B | 5q31.1 | Intracellular chylomicron trafficking |
4.3.1 Carbohydrate Malabsorption and Disaccharidase Deficiencies
Carbohydrate absorption begins with the breakdown of complex carbohydrates by salivary and gastric enzymes into oligosaccharides, which are then hydrolyzed to monosaccharides by specific disaccharidases located at the enterocyte brush border. Internalization of the hexose molecule is then mediated by “active” glucose absorption across the brush-border membrane by the Na+− dependent sodium/glucose cotransporter (SGLT1). Absence of SGLT1 results in glucose–galactose malabsorption (Hediger et al. 1989, 1995). There are also facilitative hexose transporters located at the basolateral membrane. The latter have been cloned and functionally characterized in several different tissues (GLUT1, erythrocyte; GLUT2, hepatocyte; GLUT3, brain; GLUT4, muscle and fat; GLUT5, small intestine) (Corpe et al. 1999).
There are four major disaccharidase enzymes: sucrase–isomaltase, glucoamylase–maltase, trehalase, and lactase–phlorizin hydrolase (or lactase-glycosylceramidase) (Auricchio and Troncone 2000). Sucrase–isomaltase is responsible for the degradation of dietary sucrose. Glucoamylase–maltase hydrolyses starch into small polymers. Lactase–phlorizin hydrolase degrades lactose, the main carbohydrate in milk. Trehalose occurs mainly in mushrooms. Trehalase deficiency is extremely rare, encountered mainly in Greenland natives and in Finns (Gudmand-Hoyer et al. 1988). The expression of these enzymes on the intestinal brush border appears to follow a time-dependent sequence. Lactase–phlorizin hydrolase, highly expressed at birth, declines in early life in most people (though it persists through adult life in Caucasians) (Welsh et al. 1978). Sucrase–isomaltase, undetectable at birth, reaches adult levels in the first few months of life. These changes coincide with the switch from milk to a solid diet.
The clinical picture of congenital disaccharidase deficiency and carbohydrate malabsorption is an osmotic diarrhea due to the unabsorbed solute in the ileum. The more rapid transit through the GI tract in the child results in a more severe diarrhea than in the adult. Symptoms are also related to factors such as residual enzyme activity, the amount of fed carbohydrate, and the degree of fermentation of the unabsorbed carbohydrate by colonic bacteria. The appearance of the small intestine in these cases is usually unremarkable, and the diagnosis arrived at by the determination of disaccharidase activities in homogenates of small bowel biopsies or by breath testing. Congenital sucrase–isomaltase deficiency is an autosomal recessive disorder occurring mainly in individuals of European descent, in whom the prevalence of heterozygosity reaches 2–9 % (Treem 1995). A diversity of different mutations underlies clinical heterogeneity, with the presentation of disease ranging from osmotic diarrhea, dehydration, and failure to thrive in infants to much milder symptoms in adults, in whom the diagnosis may be considerably delayed (Treem 1996). In most cases the disease becomes apparent when the child starts to consume fruits and juices (Canani and Terrin 2011). Lactose intolerance can be divided into three different groups: the rare congenital lactase deficiency characterized by complete absence of the enzyme from birth, manifesting as a severe osmotic diarrhea and dehydration; the very common hypolactasia which is the most common form of lactose malabsorption and which seems to be a consequence of a genetically mediated downregulation of the lactase enzyme (Campbell et al. 2009); and secondary lactase deficiency consequent to mucosal injury as in infectious diarrhea, celiac, or Crohn’s disease (Schulzke et al. 2009). Maltase–glucoamylase deficiency is as yet poorly defined and which appears closely related to sucrase–isomaltase deficiency (Canani and Terrin 2011).
Glucose–galactose malabsorption is a rare autosomal recessive disorder resulting from a mutation in the gene for SGLT 1 (also known as sodium/glucose cotransporter, solute carrier family 5, member 1, SLC 5A1), located on chromosome 22, which can cause severe chronic diarrhea (Xin and Wang 2011). Congenital fructose malabsorption is the result of a mutation in the gene for the hexose transporter isoform, GLUT5, located on chromosome 1 (Burant et al. 1992; Davidson et al. 1992). In clinical practice, malabsorption of fructose and that of a closely related sugar, sorbitol, may also result from overfeeding with juices containing high levels of these sugars.
4.3.2 Lipid Trafficking Disorders
Most clinical disorders of fat malabsorption result from either severe liver disease, pancreatic disease (such as cystic fibrosis), or extensive ileal resection (as in Crohn’s disease) with loss of the enterohepatic circulation of bile acids. Intestinal biopsies play a limited role in the diagnosis of these disorders. However, primary abnormalities involving abnormalities of fat transport within the enterocyte, though much less frequent, can result in a characteristic vacuolization of the enterocyte in intestinal biopsies.
4.3.2.1 Abetalipoproteinemia
Abetalipoproteinemia is an autosomal recessive disorder characterized by the absence of apoB-containing lipoproteins. The molecular basis for the defect is a mutation of the gene coding for microsomal triglyceride transfer protein (MTP), located on chromosome 4q22 (Table 4.4) (Wetterau et al. 1992). MTP is responsible for assembly of lipoprotein particles, and for the proper folding of ApoB, preventing its premature degradation (Berriot-Varoqueaux et al. 2000). Fatty acids within intestinal cells thus cannot be exported as chylomicrons. Patients have diarrhea and fat malabsorption usually appearing within the first few months of life, with acanthocytosis, and deficiencies in fat-soluble vitamins resulting in retinitis pigmentosa and neurologic symptoms. There is clinical heterogeneity, however, with signs and symptoms presenting in older patients in a significant proportion of cases. Serum levels of cholesterol and triglycerides are typically low and do not rise after a fatty meal.
4.3.2.1.1 Pathology
Villus morphology is generally preserved; characteristic multi-vacuolated fat-filled enterocytes are noted on intestinal biopsies of fasting patients, which on electron microscopy are irregular in size and generally non-membrane bound (Fig. 4.9). No lipid is noted in the extracellular space. Hepatic biopsies in these cases typically reveal steatosis, with numerous non-membrane-bound lipid droplets noted in the hepatic cytoplasm (Avigan et al. 1984). Fibrosis evolving to cirrhosis has been reported in occasional patients (Black et al. 1991).
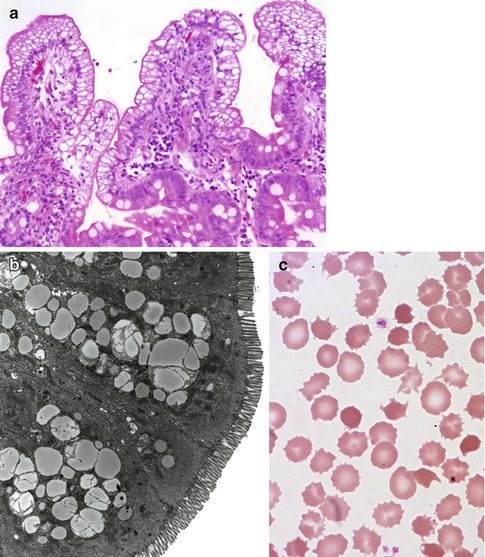
Fig. 4.9
Abetalipoproteinemia. (a) Duodenal biopsy from a 9-month-old male with severe failure to thrive and elevated liver enzymes. Villus structure is well preserved. There is diffuse vacuolization of the enterocytes (H+E, ×200). (b) Lipid filled supranuclear vacuoles of varying sizes are noted on ultrastructural examination (thin sections courtesy of Dr Pierre Brochu, Hopital Ste-Justine, Montreal). (c) Same patient as (a); acanthocytes and burr cells are observed on the blood smear
4.3.2.2 Hypobetalipoproteinemia
Hypobetalipoproteinemia is an autosomal dominant disorder due to a mutation in the apoB gene located on chromosome 2, leading to a truncated apoB protein (Peretti et al. 2010). Homozygous patients have a clinical and histologic phenotype essentially indistinguishable from abetalipoproteinemia, whereas heterozygous patients have only a mild phenotype.
4.3.2.3 Chylomicron Retention Disease
Chylomicron retention (Anderson) disease) is similar to abetalipoproteinemia in its gastrointestinal manifestations and impact on growth, though acanthocytosis is usually absent and neurologic and ocular abnormalities are much less severe. Also, in contrast to abetalipoproteinemia, serum fasting triglyceride levels are normal and hypocholesterolemia is less marked. The causative gene, SAR1B, codes for a GTPase associated with coat protein carriers involved in endoplasmic reticulum to Golgi transport, particularly in chylomicron and LDL transport (Jones et al. 2003).
4.3.2.3.1 Pathology
The endoscopic and histologic features are essentially similar to abetalipoproteinemia. Endoscopy finds a white duodenal mucosa with normal esophageal and gastric mucosa. Fat-filled enterocytes are noted primarily in the upper portion of the villi (Fig. 4.10). Villus morphology is preserved though some patients may demonstrate mild villus atrophy. It has been noted that the extent of vacuolization may vary along different villi in the same patient (Peretti et al. 2010). Lipid vacuoles fill the enterocytes on electron microscopy; some observers have noted that the lipid droplets in chylomicron retention disease appear to be more uniform and membrane-bound than those in abetalipoproteinemia (Boldrini et al. 2001).
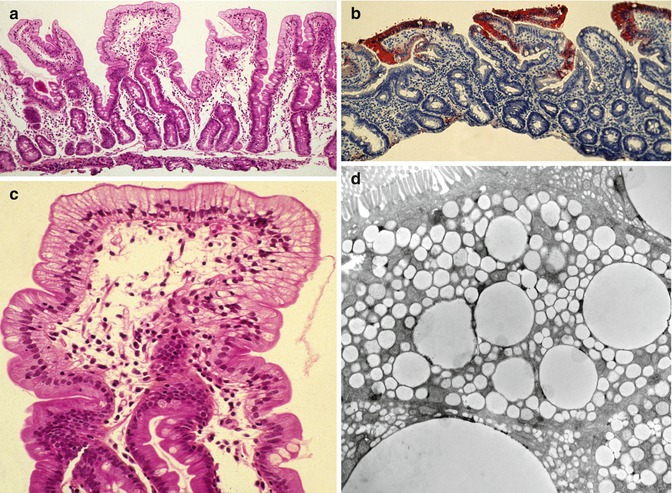
Fig. 4.10
Chylomicron retention disorder. (a) Duodenal biopsy showing well-preserved villi with diffuse vacuolization of the epithelium (H+E, ×100). (b) The vacuoles are positively stained with Oil Red O due to the accumulation of neutral lipids (Oil Red O, ×100). (c) Higher power view of (a) showing diffuse vacuolization especially along the upper portion of the villi. (d) Electron microscopy confirms accumulation of variably sized lipid droplets in the enterocytes (Case courtesy of Dr. Pierre Brochu, Hopital Ste-Justine, Montreal, Canada)
Enterocyte vacuolization may result from several other entities (Table 4.5). Minor degrees of enterocyte vacuolization, due to a recent feed, for example, are common in intestinal biopsies in infants; in these cases, vacuolation is neither as marked nor as diffuse as in abetalipoproteinemia or chylomicron storage disease (Fig. 4.11). In addition, lipid droplets are present in the intercellular spaces and lacteals after feeding but are absent in these spaces in disorders causing impaired lipid transport (Variend et al. 1984b).
Table 4.5
Entities associated with enterocyte steatosis
Abetalipoproteinemia |
Homozygous hypobetalipoproteinemia |
Chylomicron retention disease (Anderson’s disease) |
Protein calorie malnutrition |
Gluten-sensitive enteropathy |
Diabetes mellitus type I |
Fig. 4.11
Minor degrees of enterocyte vacuolization are not uncommon after recent ingestion of fat, as shown here in a duodenal biopsy from an 18-year-old male investigated for reflux disease (H+E, ×200)
4.3.3 Other Transport Disorders
4.3.3.1 Amino Acids
Disorders of amino acid transport rarely feature prominent gastrointestinal manifestations, except for lysinuric protein intolerance (LPI), which results from mutations in the SLCA7 gene which codes for the light chain subunit of the heterodimer amino acid transporter at the basolateral membrane of intestinal and renal epithelial cells (Chillaron et al. 2001). Initially described in Finland, but now recognized worldwide, LPI clinically manifests as failure to thrive with vomiting and diarrhea. Hepatosplenomegaly, hematological anomalies, and neurological involvement, including hyperammonemic coma, are recurrent clinical features. Ornithine and arginine, urea cycle intermediates that fail to exit from intestinal epithelium in LPI, result in a urea cycle dysfunction with hyperammonemia and alteration in mental status. Two major complications, pulmonary alveolar proteinosis and renal disease, are increasingly observed in LPI patients (Sebastio et al. 2011; Sperandeo et al. 2008). The condition is recognized by the markedly elevated urinary levels of lysine and other dibasic amino acids; DNA testing may be required for confirmation of the diagnosis. A 5-year-old boy was reported with chronic diarrhea and a flat gut on small intestinal biopsy and unsuccessful treatment with a gluten-free diet (Reinoso et al. 1998). Other complications associated with this disorder include lupus, hemophagocytic lymphohistiocytosis (Bader-Meunier et al. 2000), and sudden infant death (de Klerk et al. 1996).
4.3.3.2 Electrolytes and Trace Elements
The biology of intestinal ion transport is a complex field with disorders ranging from rare selective deficiencies to multisystem disease such as cystic fibrosis. The understanding of solute transport is rapidly expanding as the individual transporter molecules that form part of the Solute Carrier Family (SLC) class of proteins are cloned and functionally characterized (Kere and Hoglund 2000). Those defects presenting primarily with severe diarrhea include congenital chloride diarrhea and congenital sodium diarrhea. Intestinal biopsy in these cases has been reported as normal or showing only mild partial villus atrophy (Keller et al. 1990). Congenital chloride diarrhea is caused by mutations in the solute-linked carrier family 26 member A3 (SLC26A3) gene. It manifests as a life-threatening congenital diarrhea. Long-term manifestations include renal disease, spermatoceles, and male subfertility (Canani et al. 2010). Congenital sodium diarrhea results in hyponatremia and metabolic acidosis. The syndromic form of this disorder has been linked to mutations in SPINT2, also characterized by choanal and anal atresia, hypertelorism, double kidney, cleft palate, and digital anomalies (Canani and Terrin 2011).
Acrodermatitis enteropathica is an autosomal recessive disorder resulting from a defect in intestinal absorption of zinc. Mutations in a candidate gene, ZIP4, result in defective epithelial transport in the intestine and kidney (Canani and Terrin 2011). There is a characteristic clinical picture of diarrhea and acral and orificial skin lesions. Rodin and Goldman found pancreatic islet hyperplasia, absence of the thymus and of germinal centers, with plasmacytosis of the spleen and lymph nodes in autopsies (Rodin and Goldman 1969). Inclusion bodies in Paneth cells have been reported in patients with this disorder (Suzuki et al. 1979; Variend et al. 1984a). A decrease in villus height is noted in animal models of zinc deficiency, which is corrected by zinc supplementation (Southon et al. 1986). Interference with chylomicron development and function has been noted (Koo et al. 1985). The disease is correctable by oral zinc supplementation. Severe zinc deficiency is also seen in other disorders such as the Cronkhite–Canada syndrome (Bacher et al. 1997).
Menkes disease is an X-linked disorder which results from a defect in intestinal absorption of copper. Affected patients present characteristic hair shaft anomalies (pili torti), cerebral degeneration, hypopigmentation, abnormal bones, vomiting and diarrhea, and occasional protein-losing enteropathy. The disorder maps to Xq12-q13 and is caused by mutations in the ATP7A gene, which maps to Xq12-13, and encodes a copper-transporting P1B-type ATPase, part of a family of membrane proteins responsible for cation transport across membranes (Moller et al. 2009; Vulpe et al. 1993).
4.3.3.3 Vitamins
Congenital vitamin B12 transport disorders include congenital intrinsic factor deficiency, a selective absence of intrinsic factor without any gastric anomaly; congenital pancreatic insufficiency, of various causes; and congenital selective vitamin B12 malabsorption, the latter an autosomal recessive disorder characterized by megaloblastic anemia, hepatosplenomegaly, vomiting, diarrhea, and proteinuria, due to mutations in the gene CUBN, which codes for an ileal cell surface receptor protein, Cubilin (Storm et al. 2011). None of these disorders results in any significant histologic changes in intestinal biopsies.
A rare congenital disorder of intracellular vitamin B12 metabolism belonging to the cobalamin C (CblC) has also been described in patients who presented with diarrhea, renal failure, and systemic thromboemboli in association with a striking gastric atrophy (Russo et al. 1992). Protein-losing enteropathy responsive to hydroxocobalamin treatment has been reported in one patient with this disorder (Ellaway et al. 1998), presumably on the basis of the gastric anomaly.
4.3.3.4 Bile Acids
Heubi and colleagues (1982) described a severe refractory diarrhea due to a primary disorder of bile acid absorption; intestinal biopsies were normal. A defect in the gene coding for an ileal Na+-dependant bile acid transporter, ASBT (SLC10A2), has been found in this disorder (Oelkers et al. 1997). It is worth noting that bile acid–related diarrheas are much more commonly secondary to chronic pancreatic insufficiency (Bruno et al. 1995) or to loss of ileal surface, as in short gut syndrome due to necrotizing enterocolitis (Ohkohchi et al. 1997), or extensive ileal resection in Crohn’s disease (Harewood and Murray 1999; Stelzner et al. 2001).
4.4 Congenital Defects of Intestinal Epithelial Differentiation
4.4.1 Microvillus Inclusion Disease (MVID)
4.4.1.1 Clinical Presentation and Genetics
Initially described by Davidson et al. in 1978 (Davidson et al. 1978) and subsequently recognized worldwide, MVID is an autosomal recessive disease characterized by refractory secretory diarrhea usually occurring within the first week of life, though a late-onset form may manifest in the first few months of life (Ruemmele et al. 2006). Investigation of clusters of cases within the Navajo population identified mutations in the gene MYO5B (Erickson et al. 2008). Homozygosity mapping of an extended Turkish kindred allowed Muller and colleagues to locate the gene locus to chromosome 18q21 (Muller et al. 2008). MYO5B codes for myosin Vb, part of a family of proteins responsible for actin-dependent organelle transport and regulation of endosome recycling (Muller et al. 2008). Mutations in the MYO5A gene cause Griscelli syndrome, an immunodeficiency disorder also characterized by abnormal transfer of melanin granules to keratinocytes (Muller et al. 2008). Patients with MVID are dependent on total parenteral nutrition (TPN), though improved survival may require small bowel transplantation (Halac et al. 2011).
4.4.1.2 Pathology
Small bowel biopsies are characterized by usually severe villus atrophy, mild or moderate crypt hyperplasia, and a variable degree of inflammation in the lamina propria but without crypt destruction (Fig. 4.12). The diagnosis may be strongly suspected on paraffin-embedded sections by the absence of a distinct brush border using the periodic acid–Schiff (PAS) stain and by the presence of PAS-positive diastase-resistant densities at the apex of the enterocytes (Fig. 4.13a, b). Similar observations are noted using immunohistochemical staining for alkaline phosphatase and anti-CD 10 (Fig. 4.13c, d), a membrane-associated neutral peptidase (Groisman et al. 2002). More recently, similar results using antibodies directed against anti-Rab 11 a, a small guanosine triphosphatase (GTPase) protein on the surface of recycling endosomes, has provided further evidence that MVID is a disorder of apical plasma membrane recycling (Talmon et al. 2011). The pathognomonic ultrastructural features include absent or small stubby microvilli and vesicular structures located toward the apex of the enterocytes containing microvilli and granules containing dense amorphous material (Fig. 4.14). Microvillus inclusions have also been reported in the colon, gallbladder, and renal tubular epithelium in these patients (Cutz et al. 1997). A variety of different ultrastructural features has also been noted in patients with this disorder, and finding the “typical” inclusions may require a prolonged search (Iancu et al. 2007a). “Atypical” inclusions include the presence of numerous small electron-lucent vesicles and accumulation of osmiophilic vermiform structures (Iancu et al. 2007b) (Fig. 4.14). Cases with these features have been previously reported as MVID with atypical features or as microvillus dystrophy (Mierau et al. 2001; Raafat et al. 1994). Genetic analysis will surely help in defining whether the ultrastructural and sometimes clinical variability represent different entities or variations in the spectrum of manifestations of a single disorder, especially given the enormous prognostic and therapeutic implications.
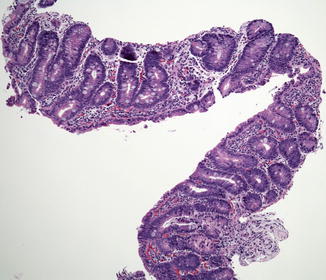
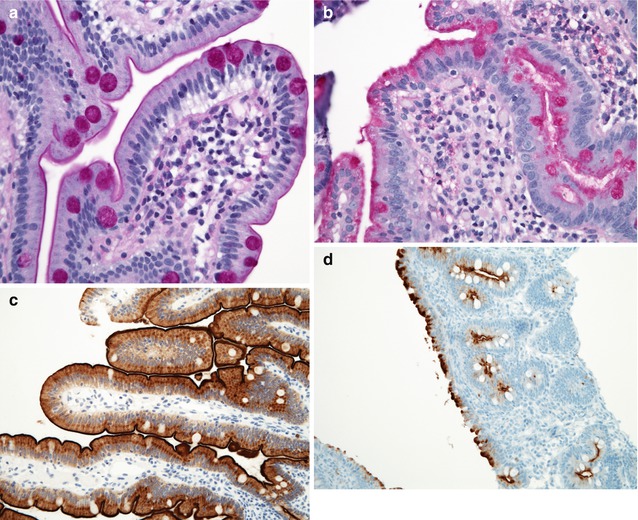
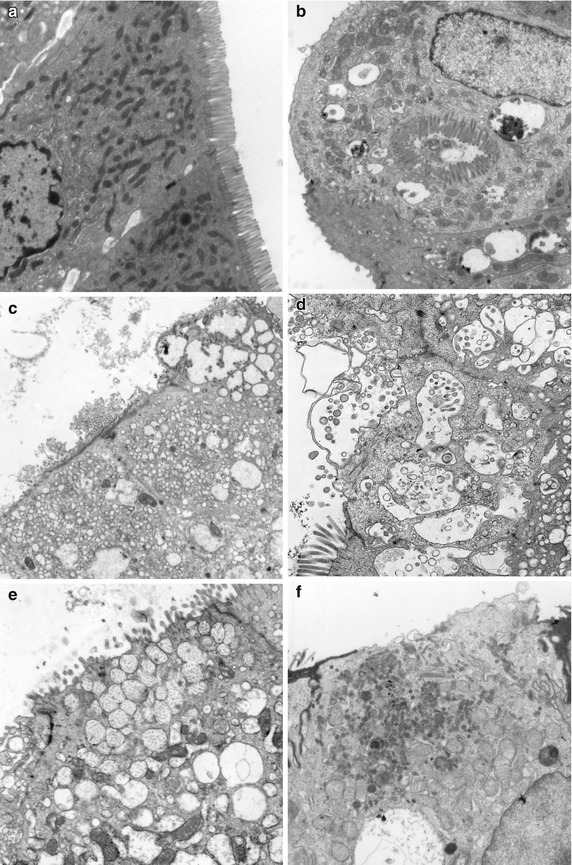
Fig. 4.12
Microvillus inclusion disease (MVID). Biopsy from an 18-day-old boy shows total villus atrophy with normoplastic crypts and no significant inflammation (H+E, ×100)
Fig. 4.13
Microvillus inclusion disease. (a, b) PAS stain. The panel on the left (a) shows a normal brush border appearing as a thin distinct magenta line along the apical border of the enterocytes. Note the normal goblet cell staining. Contrast with (b), on the right, from a case of MVID, showing a lack of a distinct brush border and a diffuse positivity in the apical portion of the enterocyte cytoplasm. Goblet cells are also present (H+E, ×40). (c) Immunostaining for CD10 reveals a linear dense positivity along the brush border of a normal duodenum (avidin–biotin immunoperoxidase, ×400). (d) By contrast, diffuse periapical cytoplasmic staining is noted in a case of MVID (same case as Fig. 4.12, avidin–biotin immunoperoxidase, ×200)
Fig. 4.14
Microvillus inclusion disease. Variety of ultrastructural features. (a) Normal duodenum with well-preserved microvilli. (b) Same case as Fig. 4.12. Enterocytes denuded of a brush border with a “typical” inclusion containing profiles of microvilli. Note also the presence of numerous surrounding vesicles containing dense amorphous material. (c) In this case from a 1 month-old female, denuded enterocytes contain numerous lucent vacuoles in the periapical cytoplasm. Several vesicles containing profiles of microvilli are noted in the enterocyte in the top right of the picture. (d) Numerous vesicles containing microvilli in profile or cross section are noted with blebbing of the cytoplasmic surface. (e) This case from a 1-year-old boy is characterized by numerous periapical vesicles containing small vermiform bodies. (f) Another case from a 2-month-old girl shows a denuded enterocyte containing numerous immature microvilli in the periapical cytoplasm that do not appear to be enclosed in vesicles (uranyl-lead, (a–c, f ×7000; d, e ×14,000)
4.4.2 Congenital Tufting Enteropathy (CTE, Epithelial Dysplasia)
4.4.2.1 Clinical Presentation and Genetics
First described by Reifen et al. (Reifen et al. 1994), patients with tufting enteropathy present in the neonatal period with a watery diarrhea. Prenatal history is uneventful and the disease appears to be inherited in an autosomal recessive fashion, as suggested by the finding of other affected siblings and frequent parental consanguinity. The incidence is estimated at 1/50,000–100,000 live births in Europe and appears to be more frequent in patients of Arabic origin (Goulet et al. 2007). Cases of tufting enteropathy have been described in association with other malformations, including facial dysmorphism, micrognathia and anal imperforation (Abely et al. 1998), and choanal atresia and hair abnormalities (Bird et al. 2007). Superficial punctate keratitis and conjunctival erosions have been observed in about 50 % of patients (Roche et al. 2010). In addition, an association has been reported between tufting enteropathy and autoimmune phenomena, including autoimmune anemia and thrombocytopenia (El-Matary et al. 2007) and chronic arthritis (Al-Mayouf et al. 2009). Using single nucleotide polymorphism (SNP) analysis in an affected kindred, Sivagnanam et al. identified the epithelial cell adhesion molecule gene (EPCAM), located on chromosome 2p21, as the causative gene (Sivagnanam et al. 2008). EpCAM belongs to a family of cell adhesion receptors, associates with tight junction proteins, and is responsible for cell–cell interaction by recruiting actin filaments to sites of contact (Carpenter and Red Brewer 2009).
A probably related disorder resulting from the absence of intestinal epithelial α6β4 integrin has been described. The salient features are severe watery diarrhea in the first 2 weeks of life in association with pyloric atresia, with extensive sloughing of the epithelium observed on biopsies (Lachaux et al. 1999). Diarrhea has also been observed in cases of pyloric atresia–junctional epidermolysis bullosa, in which mutations in the genes coding for laminin-5 and α6β4 integrin have been described (Korber and Glasson 1977).
4.4.2.2 Pathology
The histologic hallmarks are severe villus atrophy with the formation of “tufts” of rounded, tear-drop-shaped enterocytes which appear to shed into the lumen. There may be a mild increase in the lamina propria inflammatory cells, but IELs do not appear to be significantly increased. The brush border is normal by PAS staining, and electron microscopic findings are nonspecific. Dilated crypts have also been described. Epithelial abnormalities typically vary over time and may be subtle or absent in the first biopsy (Fig. 4.15). Abnormalities in immunohistochemical staining for basement membrane laminin and heparan sulfate proteoglycan (Cutz et al. 1997), ultrastructural irregularities of desmosomes, and abnormal distribution of α2β1 integrin (Kagitani et al. 1998) have been observed in patients with this disorder. Intestinal features resembling CTE have been noted in a knockout mouse model for transcription factor Elf3 (Ng et al. 2002). A very helpful feature is the finding that immunohistochemistry of patient intestinal tissue reveals decreased expression of EpCAM (Salomon et al. 2011) (Fig. 4.16).
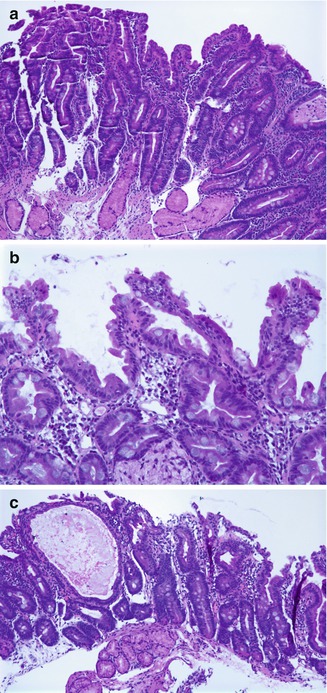
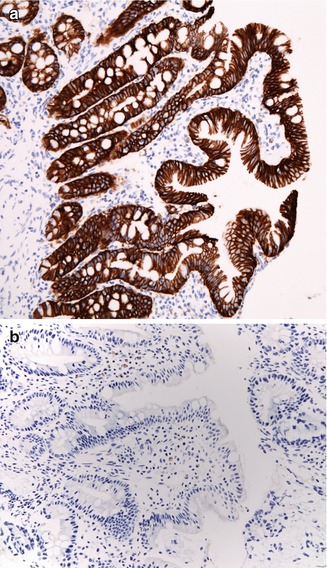
Fig. 4.15
Tufting enteropathy. (a) Duodenal biopsy at 2 months of age shows villus atrophy and some surface irregularity but no definite tufting. (b) Biopsy at 4 months of age shows marked tufting of the epithelial surface. (c) Biopsies performed at 5 months of age revealed, in addition to areas of tufting, cystically dilated crypts (H+E, ×100)
Fig. 4.16
Tufting enteropathy. Immunostaining for MOC 31, an antibody against EpCAM. (a) Normal staining of colonic surface and crypt epithelium with a membranous accentuation. (b) Absence of staining in a case of tufting enteropathy (immunoperoxidase with hematoxylin counterstain, ×200)
The disease appears to have a variable clinical course with some patients reported to have been weaned off TPN (Lemale et al. 2011). The report of a patient with similar clinical and biopsy features who was found to harbor a mutation in SPINT2, associated with congenital sodium diarrhea, suggests there may be phenotypic overlap between CTE and other congenital diarrheas (Sivagnanam et al. 2010).
4.4.3 Enteroendocrine Cell Dysgenesis (ECD, Enteric Anendocrinosis)
4.4.3.1 Clinical Presentation and Genetics
ECD is a recently described autosomal recessive disorder associated with a congenital absence of enteroendocrine cells in the small and large bowel secondary to mutations in the NEUROG3 gene, located on chromosome 10q21.3, required for differentiation of epithelial cells to the endocrine phenotype (Wang et al. 2006). Affected patients are characterized by a congenital diarrheal syndrome with profound malabsorption of all nutrients from birth. Patients also typically develop insulin-dependent diabetes, some during infancy and others later in childhood (Pinney et al. 2011; Wang et al. 2006). Pancreatic exocrine function is normal. Neurogenin-3 null mice lack enteroendocrine cells (Bjerknes and Cheng 2006) and are severely diabetic as neurogenin-3 is also required for pancreatic endocrine development (Gradwohl et al. 2000).
4.4.3.2 Pathology
The intestinal biopsies may be normal or reveal villus atrophy, the severity of the changes perhaps related to different mutations in the NEUROG 3 gene (Pinney et al. 2011) (Fig. 4.17). The brush border is normal, and goblet and Paneth cells are present. Inflammatory cells in the lamina propria do not appear significantly increased, and there is no crypt-destructive process. Electron microscopy confirms the presence of microvilli and the absence of microvillus inclusions (Cortina et al. 2007). The characteristic absence of enteroendocrine cells in the small bowel and colon is confirmed by immunohistochemistry for chromogranin A (Fig. 4.17). Patients with autoimmune polyglandular syndrome I may have absent or markedly reduced enteroendocrine cells, which may be transient (Posovszky et al. 2012; Hogenauer et al. 2001) – vide infra. Loss of enteroendocrine cells may be observed in patients with autoimmune enteropathy, along with loss of goblet and Paneth cells.
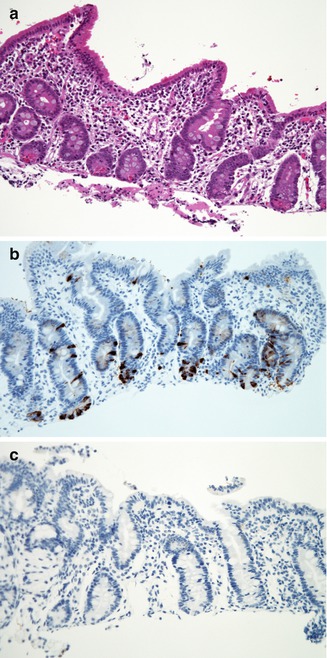
Fig. 4.17
Enteroendocrine cell dysgenesis. (a) Duodenal biopsy from a 10-month-old female who presented with diarrhea shortly after birth and insulin-dependent diabetes at 10 months. There is partial villus atrophy, mild crypt hyperplasia, and no significant inflammation (H+E, ×200). (b) Normal distribution of enteroendocrine cells in the duodenum as revealed by immunostaining for chromogranin (avidin–biotin peroxidase, X200). (c) Same case as (a), with complete absence of immunoreactivity for chromogranin (avidin–biotin peroxidase, ×200)
4.4.4 Syndromic Diarrhea (Tricho-hepato-enteric Syndrome)
4.4.4.1 Clinical Presentation and Genetics
Though not known to be a disorder of intestinal differentiation, syndromic diarrhea and the tricho-hepato-enteric syndrome are discussed here because they enter into the differential diagnosis of early-onset diarrhea. Girault et al. (Girault et al. 1994) described a group of patients with dysmorphic features, consisting of a prominent forehead, broad nose, hypertelorism, and wooly, easily removable abnormal hair (trichorrhexis nodosa), in association with intractable diarrhea and immunodeficiency, which they termed syndromic intractable diarrhea, because of the constellation of extraintestinal manifestations. Most of the patients reported were of Middle Eastern origin, and there was parental consanguinity and a history of similarly affected siblings. The immunodeficiency was characterized by impaired T-cell and antibody responses despite normal immunoglobulin levels. Infants with similar clinical features were also reported as the tricho-hepato-enteric syndrome (THE) (Stankler et al. 1982; Verloes et al. 1997), in which thin sparse hair (trichomalacia) was seen in association with hypertelorism and chronic diarrhea and a history of premature delivery with intrauterine growth retardation. In addition, the cases described by Verloes also presented with neonatal hemochromatosis, characterized by liver failure, cirrhosis, and multivisceral iron deposition (Verloes et al. 1997). The phenotypic similarity between THE and the cases described by Girault led Fabre and colleagues to suggest that these manifestations represented a spectrum of the same disorder (Fabre et al. 2007). This appeared to be confirmed when mutations in TTC37, described in cases of THE (Hartley et al. 2010), were also observed in patients with the features of “syndromic diarrhea” (Fabre et al. 2011). TTC37 maps to chromosome 5q14.3–5q21.2 and codes for a tetratricopeptide repeat protein, thespin, whose function is as yet uncharacterized (Hartley et al. 2010).
4.4.4.2 Pathology
Intestinal biopsies have revealed variable villus atrophy without a conspicuous increase in inflammatory cells and without specific features. Serially obtained biopsies suggest that villus atrophy can improve with time and that the inflammatory infiltrate is not consistent. The hair is described as wooly with microscopic features of trichorrhexis nodosa (Fig. 4.18). Hepatic involvement is inconsistent. Hartley et al. also observed ultrastructural platelet abnormalities consisting of reduced α-granules and abnormal lipid inclusions and canalicular system (Hartley et al. 2010).
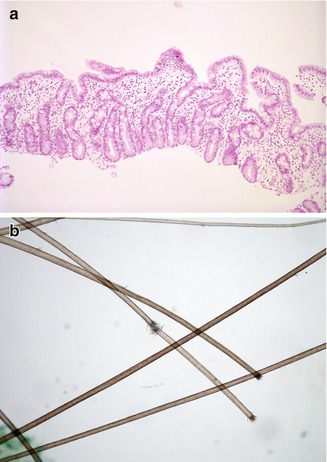
Fig. 4.18
“Syndromic” diarrhea. (a) Duodenal biopsy from a 2-month-old boy shows partial villus atrophy, mild crypt hyperplasia, and lamina propria inflammation. (b) Trichorrhexis nodosa. Breaks in the hair shafts resulting in “brush ends”
We have observed trichorrhexis nodosa in a 3-month-old girl presenting with severe diarrhea and an immunodeficiency best characterized as common variable immunodeficiency. Hair abnormalities can be seen in association with chronic diarrhea in other disorders, typically in Menkes disease, zinc deficiency, and lysinuric protein intolerance.
4.5 Immune-Mediated Disorders
4.5.1 Autoimmune Enteropathy (AIE)
4.5.1.1 Clinical Manifestations and Genetics
Autoimmune enteropathy is probably the most frequent disorder leading to infantile intractable diarrhea (Montalto et al. 2009). Most cases occur in infancy or the first year of life. There is a strong male preponderance, family history of other affected siblings, frequent extraintestinal involvement, and various circulating autoantibodies (Russo et al. 1999b; Tsuda et al. 2010). Although the IPEX syndrome (immune dysregulation, polyendocrinopathy, enteropathy, X-linked, reviewed in Chap. 5 of this textbook, Sect. 5.1.3.5) due to mutations of FOXP3 is the most well-characterized form of AIE, the latter is clearly a heterogeneous entity which has been described in girls, older children, and even in adults. In fact, about 50 % of patients, including girls, with otherwise typical clinical manifestations of IPEX have no detectable mutations of FOXP3 (Torgerson and Ochs 2007). The term “IPEX-like” has been suggested to designate those cases (Yong et al. 2008). The clinical picture in infants is usually dominated by a secretory diarrhea, dermatitis, and various endocrine disorders. Moreover, these patients also suffer from numerous complications, mainly infectious, related to loss of the functional gut barrier, central lines, underlying immunodeficiency, and immunosuppressive therapy. In adults, the clinical picture appears to be that of celiac disease unresponsive to gluten withdrawal (Akram et al. 2007; Carroccio et al. 2003; Corazza et al. 1997; Daum et al. 2003; Mais et al. 1999; Marthinsen et al. 2008; Mirakian et al. 1997; Mitomi et al. 1998). Corazza et al. reported four adult women with refractory celiac disease, none of whom had had malabsorptive symptoms in childhood, thus confirming an adult onset (Corazza et al. 1997). Akram et al. reported on a series of 15 (7 females) patients, aged 42–67 years, in whom celiac disease was excluded by lack of response to gluten-free diet or absence of the celiac disease susceptibility HLA genotypes. All patients had protracted diarrhea, weight loss, and malnutrition (Akram et al. 2007).
4.5.1.2 Pathology
Intestinal biopsies in most of the reported cases are characterized by severe villus atrophy, crypt hyperplasia, and a mixed, lymphocyte-predominant inflammatory infiltrate of the lamina propria. Concomitant colitis and gastritis are present in the majority of cases, and in the absence of a diagnosis of IPEX, an early form of chronic inflammatory bowel disease is a consideration. The histopathologic features of IPEX-like cases are essentially indistinguishable from cases with mutations of FOXP3. Marked inflammatory destruction of intestinal crypts with extensive apoptosis is a feature noted in many of the cases, similar to those noted in intestinal graft-versus-host disease, and suggests an abnormal immune-mediated attack against intestinal epithelium (Fig. 4.19a). A characteristic histologic feature is absence or severe reduction in goblet and Paneth cells, related to either immune-mediated destruction or rapid epithelial regeneration that does not allow complete histologic differentiation. However, there appears to be marked variation in the histologic features, with some cases showing milder degrees of intestinal damage. In contrast to cases of gluten-sensitive enteropathy with flat villi, intraepithelial lymphocytes tend to be relatively few in number.
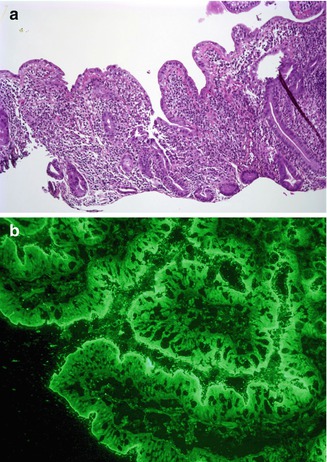
Fig. 4.19
Autoimmune enteropathy, IPEX like. (a) Duodenal biopsy from a 2-year-old female shows villus atrophy and a severe crypt destructive inflammatory process (H+E, ×100). (b) Indirect immunofluorescence using the patient’s serum reveals linear staining along the periapical portion of the enterocytes, consistent with the presence of circulating anti-enterocyte antibodies
4.5.1.3 Diagnosis and Treatment
One of the hallmarks of this entity, noted since the first reports of AIE, is the presence of anti-enterocyte antibodies, detected by indirect immunofluorescence using the patient’s serum on frozen sections of normal bowel. Positive fluorescence usually results in a linear pattern along the apex and basolateral border of the enterocyte (Cutz et al. 1997; Mirakian et al. 1986; Russo et al. 1999a; Savage et al. 1985; Unsworth et al. 1982) (Fig. 4.19). The antibodies are predominantly IgG and have been described as complement fixing (Mirakian et al. 1986), though IgM and IgA have also been described (Charritat and Polonovski 1987; Savage et al. 1985). Antibodies reacting against mucus or goblet cells have also been described, and intestinal biopsies in these cases have shown a marked depletion of goblet cells (Moore et al. 1995; Rogahn et al. 1999) (Fig. 4.20).
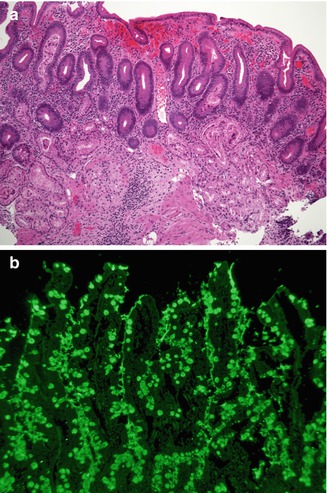
Fig. 4.20
Autoimmune enteropathy. (a) A 25-year-old female with Kabuki syndrome and long-standing diarrhea refractory to a gluten-free diet. The duodenal biopsy shows villus atrophy, crypt hyperplasia, but no significantly increased intraepithelial lymphocytes in the surface epithelium. Note the absence of Paneth and goblet cells (H+E, ×100). (b) Indirect immunofluorescence using the patient’s serum reveals positive staining within the goblet cells
The target of the anti-enterocyte antibodies is still a matter of investigation, though a 75-kilodalton (kDa) antigen reactive with autoantibody in the sera of patients with the IPEX syndrome has been reported (Kobayashi et al. 1998). This protein is expressed in the epithelial cells of the small bowel, colon, and kidney and may play a role in protein–protein interactions in the enterocyte. More than one antigen is likely to be involved. More recently, a 95-kDa antigen identified as villin has been reported in IPEX patients (Kobayashi et al. 2011).
However, the role of these antibodies in the diagnosis of AIE is debatable. These antibodies have been reported to occur after the onset of the disease and to disappear before restoration of mucosal integrity (Unsworth et al. 1982) and may thus represent an epiphenomenon without playing a causative role in mucosal damage. Similar anti-enterocyte antibodies have also been described in adult AIDS patients without digestive symptoms (Martin-Villa et al. 1993). The specificity of anti-goblet cell antibodies has also been questioned. Anti-goblet cell antibodies have been detected in patients with chronic inflammatory bowel disease (Crohn’s and ulcerative colitis) and their first-degree relatives (Folwaczny et al. 1997) and in a series of treated and untreated patients with celiac disease and controls (Biagi et al. 2009). Finally, the usefulness of these antibodies in the diagnosis of AIE in young infants is also probably limited, given that there is little IgG production in the first 3 months of life and that IgG detected in the infant is likely maternal in origin (Morell et al. 1972).
The differential diagnosis of AIE includes, in addition to celiac disease and graft-versus-host disease, Crohn’s disease and enterocolitis associated with various immunodeficiency states. A high index of suspicion for AIE should be present when there is a severe inflammatory crypt destructive process with villus atrophy and increased apoptosis in the base of the crypts, loss of goblet and Paneth cells, or an intestinal inflammatory process in a patient with autoimmune phenomena. Testing for circulating anti-enterocyte antibodies is useful in the older child and adult, but may be of more use limited in the young infant. Immunostaining for the FOXP3 gene product applied to formalin-fixed paraffin-embedded gastrointestinal biopsies may be useful to screen for the IPEX syndrome in young patients with severe diarrhea (Heltzer et al. 2007). Mortality in cases of AIE has been high, occurring in up to one-third of reported cases, but more frequent in the older descriptions. Diet changes and steroids alone have been largely ineffective, and the use of immunosuppressants appears to be necessary in most cases. Bone marrow transplantation may be curative in cases of IPEX (see Chap. 5). Immunosuppression is required in older children and adults (Akram et al. 2007). A good response to Sirolimus has been reported in IPEX-like cases (Yong et al. 2008).
Major differentiating features between various etiologies of severe diarrhea of early infancy are presented in Table 4.6.
Table 4.6
Differentiating features of severe diarrhea of early infancy
Microvillus inclusion disease | Tufting enteropathy | Enteroendocrine cell dysgenesis | Autoimmune enteropathy | |
|---|---|---|---|---|
Presentation | First 2 weeks | First 2 weeks | First 2 weeks | After 1 month |
Gene defect | MYO5b (18q21) | EpCAM (2p21) | NEUROG 3 (10q21.3) | FOXP3 (Xp11.23) in IPEX syndrome |
Extraintestinal disease | Low GGT cholestasis post-bowel transplantation | Dysmorphism; autoimmune diseases (arthritis) | Insulin-dependent diabetes | Polyendocrinopathies |
Anti-enterocyte antibodies | No | No | No | Yes |
Villus atrophy | Yes | Variable | Variable | Variable |
Surface epithelium | Absent brush border | Tufting and desquamation | Normal | Normal or atrophic |
Lamina propria inflammation | minimal | variable | Minimal | Usually increased |
4.5.2 Autoimmune Polyendocrine Syndrome (APS 1)
Also known as autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED), this is an autosomal recessive disorder with heterogeneous clinical manifestations. It is caused by mutations in the AIRE gene, which codes for a transcription factor primarily expressed in medullary thymic epithelial cells where negative selection is thought to occur (Kampe 2009). The type I form is characterized by various endocrinopathies of an autoimmune nature, often beginning in childhood or early teenage years, by chronic mucocutaneous candidiasis due to a T-cell defect, starting soon after birth, and by dystrophy of ectodermal tissues (Betterle et al. 1998). Malabsorption can occur in up to 25 % of patients and appears to result from destruction of intestinal endocrine cells (Posovszky et al. 2012). A specific deficiency of cholecystokinin-producing enteroendocrine cells has been reported in one patient, with seeming reappearance of the cells when the diarrhea abated (Hogenauer et al. 2001). Small bowel biopsies show mild changes or can even be normal, in contrast with the crypt-destructive inflammation usually seen in autoimmune enteropathy.
4.5.3 Celiac Disease (CD, Gluten-Sensitive Enteropathy)
4.5.3.1 Clinical Manifestations and Diagnosis
CD is a lifelong immune-mediated systemic disorder elicited by gluten, the major protein found in wheat, barley, and rye, in genetically predisposed individuals, resulting in damage to the intestinal mucosa and a variety of clinical manifestations and antibodies. The disease is most prevalent in Western Europe and North America, with reported incidence rates of up to 1:100, but occurs worldwide (Rodrigues and Jenkins 2008). The higher incidence rates observed more recently result from the more widespread use of serologic tests in detection and screening, from greater awareness of atypical and silent forms of the disease, and from recognition of an increased incidence of GSE in patients with Down syndrome, IgA deficiency, and diabetes (Fasano 2005). Gluten is digested to the 33-amino acid alcohol-soluble gliadin, which is resistant to further enzymatic degradation. Gliadin appears to induce intestinal epithelial cells to express IL-15, which in turn leads to activation and proliferation of the characteristic intraepithelial CD8+ T lymphocytes. These become cytotoxic and kill enterocytes with surface MIC-A, an HLA class I-like protein expressed during stress. This results in further increased intestinal permeability to gliadin, followed by its deamidation by tissue transglutaminase (tTG), an enzyme normally present in the gut, forming a neoantigen. Subsequent binding of this neoantigen to HLA molecules located on antigen-presenting cells results in secretion of inflammatory cytokines and recruitment and activation of lamina propria T lymphocytes, with production of interferon-γ and further tissue damage. The principal determinants of genetic susceptibility for CD are major histocompatibility class II genes located on the short arm of chromosome 6. The HLA-DQ2 heterodimer (DQA1*0501 and DQB1*0201) is present in 95 % of patients with CD, and most of the remainder have the HLA-DQ8 heterodimer (DQA1*0301 and DQB1*0302). HLA-DQ2 homozygous individuals have a fivefold higher risk of disease development than HLA-DQ2 heterozygous individuals (Husby et al. 2012). Expression of these HLA molecules is necessary but not sufficient to cause disease as the HLA-DQ2 haplotype is present in 30–40 % of the Caucasian population. Outside the HLA region, several genes controlling the immune response have also been related to CD (Husby et al. 2012).
The classic presentation occurs in a child 6–24 months after gluten ingestion and consists of abdominal distension, diarrhea, and failure to thrive. The age at presentation of GSE appears to vary according to the age at introduction of gluten in the diet and to the quantity consumed (Ascher et al. 1997). A variety of non-gastrointestinal symptoms have also been associated with CD, including dermatitis herpetiformis, short stature, delayed puberty, dental enamel hypoplasia, osteopenia, hepatitis, and iron deficiency anemia unresponsive to iron therapy (Hill et al. 2005) (Table 4.7). Presenting symptoms in younger children may also include vomiting, abdominal distension, and failure to thrive, whereas growth retardation and delayed puberty may be major manifestations in older children. Serologic testing of children with short stature identified a prevalence rate of 8–10 % of CD (Tumer et al. 2001). The lack of “classic” symptomatology in older children and adults frequently results in delay of diagnosis (Fasano 2005). Elevated transaminase levels associated with a nonspecific hepatitis which normalize on a gluten-free diet has been reported in adults, though that association is unclear in children, as is iron deficiency anemia unresponsive to iron therapy (Bardella et al. 1995).
Table 4.7
Major clinical presentations of celiac disease
“Classic” – malabsorption, diarrhea, wasted buttocks | ||
Vomiting abdominal distension – especially in children <1 year of age | ||
Failure to thrive | ||
Iron-resistant anemia |  | Usually in older children |
Rickets | ||
Short stature | ||
Pubertal delay | ||
Personality problems | ||
Dermatitis herpetiformis | ||
Dental enamel hyperplasia | ||
Epilepsy with cerebellar calcification | ||
According to the guidelines published in 2005 by the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition (NASPGHAN), the gold standard for diagnosis remains the intestinal biopsy (Hill et al. 2005). However, a stepwise approach is recommended with serologic testing as the initial step in symptomatic patients. Testing is also recommended for their first-degree relatives and in asymptomatic patients with conditions known to be associated with celiac disease, such as Down syndrome, Turner syndrome, Williams syndrome, autoimmune thyroiditis and, selective IgA deficiency (Hill et al. 2005) (Table 4.8). In particular, routine screening of all patients with type 1 diabetes mellitus is recommended, in whom a prevalence rate of CD up to 8 % has been reported (Armstrong et al. 2012). The prevalence of celiac disease in first-degree relatives varies between 3 and 17 % (Biagi et al. 2008). One study found that histologic abnormalities were found in duodenal biopsies in up to 42 % of DQ2+ first-degree relatives, most of them characterized by a lymphocytic enteritis (Esteve et al. 2006). Initial testing is carried out while subjects are ingesting gluten. Measurements of serum IgA-tTG (tissue transglutaminase) and IgA-EMA (endomyseal antibodies) have ranges of sensitivity and specificity of 90–100 %, provided the subjects are not IgA deficient, and are considered superior to anti-gliadin antibodies for the diagnosis of CD (Rodrigues and Jenkins 2008; Giersiepen et al. 2012). For subjects with low serum IgA levels, testing with IgG-tTG and IgG-EMA is recommended. It should be noted that antibodies to tTG and EMA are more frequently falsely negative in children less than 2 years of age, in whom additional testing for anti-gliadin antibodies, or for deamidated gliadin peptides, is recommended (Maglio et al. 2010; Armstrong et al. 2012). Detecting celiac disease in individuals who are on a self- or parent-prescribed gluten-free diet (GFD) is challenging as both CD-specific antibodies and histology may normalize following GFD. In those cases, testing for HLA-DQ2 and HLA-DQ8 may be useful as celiac disease is most unlikely in those who lack these markers (Armstrong et al. 2012). The most recent published guidelines of the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN) also recommend that serologic testing positive for CD be confirmed by histology; however, contrary to North American practice, it is suggested that biopsy may be optional in those patients with CD symptomatology and very high levels of anti-tTG and anti-EMA, provided the test is repeated at a later date to avoid false-positive tests due to laboratory or labeling error (Husby et al. 2012).




Table 4.8
Conditions associated with an increased prevalence of celiac disease in childhood
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
