I. David Weiner, Charles S. Wingo
Endocrine Causes of Hypertension
Aldosterone
Recent advances in the diagnosis of aldosterone-induced hypertension have led to the recognition that primary hyperaldosteronism is more common than previously thought. Effective diagnostic strategies are available, and treatment regimens are highly efficacious.
Etiology and Pathogenesis
Aldosterone is a steroid hormone normally produced by the zona glomerulosa of the adrenal glands (Fig. 40-1). Aldosterone synthase, which is encoded by the gene CYP11B2, is the generally accepted rate-limiting enzyme in adrenal aldosterone production. Table 40-1 summarizes factors known to stimulate or to inhibit aldosterone synthesis by the adrenal gland. Aldosterone exhibits a circadian change in serum concentration, greatest in the late morning and with peak values about 50% greater than the average concentration. The factors accepted as physiologically important regulators of aldosterone production include angiotensin II (Ang II), which stimulates aldosterone production through activation of the AT1 receptor, and atrial natriuretic peptide and chronic hypokalemia, which inhibit aldosterone production.1
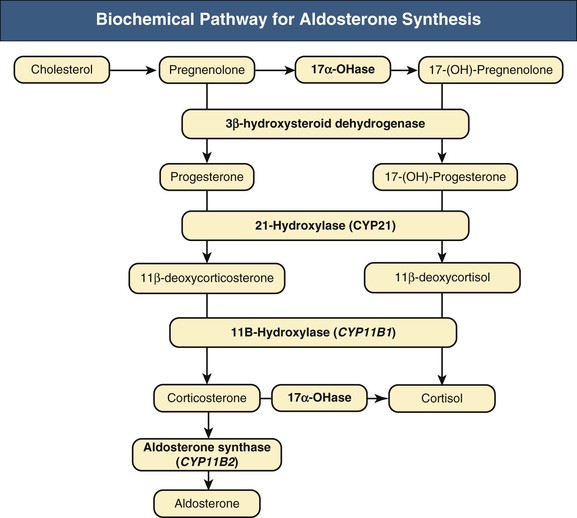
Table 40-1
Factors that regulate aldosterone release.
The stimulatory and inhibitory regulators that exert significant effects on aldosterone release under the majority of clinical circumstances are noted in italics.
| Factors that Regulate Aldosterone Release | |
| Stimulatory | Inhibitory |

Substantial advances have been made recently in our understanding of the factors that lead to primary aldosteronism. In particular, mutations in the gene for the potassium channel KCNJ5 are now recognized as a frequent cause of primary aldosteronism. Germ-line mutations cause bilateral, familial primary aldosteronism, and somatic mutations are present in about 40% of aldosterone-producing adenomas.3,4 Mutations in the genes ATP1A1 (encoding a Na+, K+-ATPase) and ATP2B3 (encoding a Ca2+-ATPase) have also been identified in about 7% of aldosterone-producing adenomas.5
A less common genetic cause is familial hypertension type 1 (FH-1), also known as glucocorticoid-remediable aldosteronism (GRA). In GRA, there is crossover between the CYP11B1 and CYP11B2 genes, resulting in a chimeric aldosterone synthase gene whose expression is regulated by adrenocorticotropic hormone (ACTH), leading to excessive aldosterone release.6,7 GRA should be considered in children or young adults with refractory hypertension or in whom there is a family history of hypertension in the same age range or history of premature hemorrhagic stroke.8 If suspected, genetic testing is the preferred diagnostic approach because of improved sensitivity and specificity over measurement of steroid metabolites or dexamethasone suppression testing.8,9 If GRA is identified, corticosteroids are administered at the minimal dose necessary to suppress ACTH release, resulting in often dramatic improvement in blood pressure (BP) control (see Chapter 49).
Aldosterone regulates BP by several mechanisms, including effects on the kidneys, vasculature, central nervous system (CNS), and other endocrine hormones (Fig. 40-2). No single effect is sufficient to explain the hypertension that occurs in primary hyperaldosteronism; together the effects explain why primary hyperaldosteronism causes refractory hypertension. Aldosterone has multiple renal effects that regulate BP. First, aldosterone stimulates renal sodium chloride (NaCl) retention by increasing expression of the thiazide-sensitive NaCl cotransporter in the distal convoluted tubule, the amiloride-sensitive epithelial sodium channel (eNaC), in the collecting duct, and the chloride-reabsorbing protein, pendrin, in the cortical collecting duct.10–12 Aldosterone also has acute effects on sodium reabsorption in these segments through mechanisms that do not require protein synthesis.13
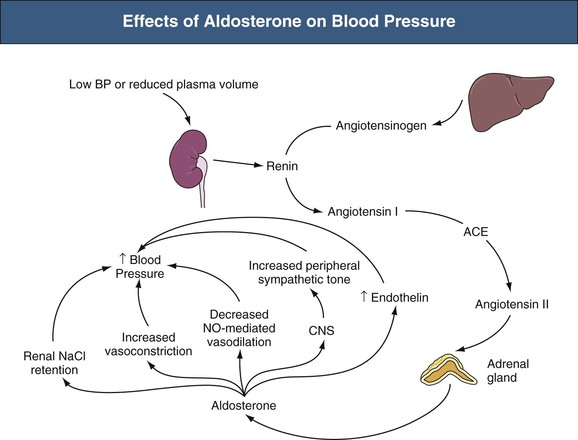
Second, aldosterone alters BP through generation of hypokalemia. The increased Na+ reabsorption enhances K+ secretion. In addition, aldosterone increases extrarenal cellular potassium uptake by stimulating the ubiquitous Na+,K+-ATPase, further decreasing extracellular potassium.14 As discussed in Chapter 9, potassium depletion raises BP through a variety of mechanisms.
Aldosterone has multiple effects on the vasculature. Aldosterone increases both basal vascular tone and vascular reactivity to circulating vasoconstrictors, including norepinephrine, epinephrine, Ang II, and vasopressin.15,16 Aldosterone decreases flow-mediated vasodilation, perhaps because of decreased nitric oxide production resulting from decreased endothelial nitric oxide synthase expression.17 In the central nervous system (CNS), aldosterone stimulates sympathetic nervous system tone, which further increases BP.18 Finally, aldosterone causes perivascular fibrosis and stimulates vascular endothelin expression.19
Aldosterone mediates its physiologic and pathophysiologic effects predominantly by activating the mineralocorticoid receptor (MR).20 The MR is located in the inactive state in the cytoplasm; aldosterone binding to MR promotes a conformational change and translocation to the nucleus, where it regulates gene transcription.
Cortisol is a naturally synthesized glucocorticoid with an affinity for MR similar to that of aldosterone, but cortisol is present in plasma at about 100-fold greater levels than aldosterone. The enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) is expressed in the aldosterone-sensitive distal nephron and collecting duct and metabolizes cortisol to cortisone, which binds to MR poorly, thereby preventing glucocorticoid-dependent MR activation. Either the genetic deficiency of or the ingestion of inhibitors of 11β-HSD can result in excessive activation of the MR and development of severe hypertension21 (see Chapter 49). Aldosterone also has nongenomic effects, but their role in mineralocorticoid-dependent BP regulation remains unclear.22,23
Primary hyperaldosteronism may result from either unilateral or bilateral adrenal disease. Typically, unilateral disease results from adenoma and bilateral disease from hyperplasia. This association is not absolute and about 10% of patients with primary hyperaldosteronism exhibit either bilateral aldosterone-producing adenoma, which may be microscopic, or unilateral hyperplasia.
Epidemiology
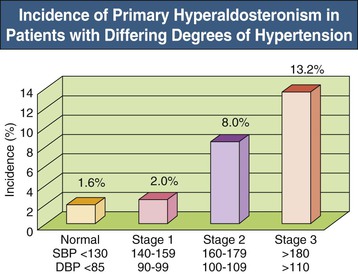
The exact incidence of primary hyperaldosteronism varies with the patient population and the diagnostic criteria used. Early studies, which only recognized severe cases, suggested that primary hyperaldosteronism was rare, with an incidence less than 1% to 2%.24 More accurate diagnosis has led to the recognition that primary hyperaldosteronism is relatively common. Patients with treatment-resistant hypertension—inadequately controlled hypertension despite treatment with three medications at appropriate dosages, including a diuretic—are highly likely to have primary hyperaldosteronism, with rates typically of 20% to 40% and as high as 67% in some studies.25 Some studies have found a 1% to 2% incidence of hyperaldosteronism in normotensive patients (see Fig. 40-3), with the incidence increasing as BP increases.26
Clinical Manifestations
Identifying patients with primary aldosteronism purely on clinical characteristics is difficult. Patients with primary hyperaldosteronism can have characteristics suggestive of secondary hypertension, such as early onset of hypertension or the need for multiple medications for BP control. Sometimes primary aldosteronism presents with either frank or easily provoked hypokalemia. Also, patients may have no distinguishing characteristics that differentiate them from individuals with essential hypertension. Figure 40-3 shows the frequency of primary hyperaldosteronism in individuals with different degrees of hypertension, and Table 40-2 summarizes characteristics of those with primary hyperaldosteronism.27
Table 40-2
Typical characteristics of patients at diagnosis of primary hyperaldosteronism.
| Typical Characteristics at Diagnosis of Primary Hyperaldosteronism | |
| Patient Factor | Measured Value |
| Gender (female/male) | 43:57 (%) |
| Age (yr) | 52 ± 1 (range 29-74) |
| Hypertension duration (yr) | 10 ± 1 |
| Number of hypertensive medications | 2.4 ± 0.1 (range 0-4) |
| Percentage requiring 3 or more medications | 54% |
| Blood pressure controlled with current medical regimen | 20% |
| Neither hypokalemic nor receiving 3 or more medications | 52% |
| Plasma aldosterone (ng/dl) | |
| <15 | 37% |
| 15-40 | 54% |
| >40 | 9% |
| Plasma renin activity (ng Ang I/ml/hr) | 0.39 ± 0.04 |
(Data from reference 27.)
Because of the wide variety of presentations, from one indistinguishable from essential hypertension to one highly suggestive of secondary hypertension, we recommend screening all patients for primary aldosteronism who have characteristics of secondary hypertension, have baseline or easily provoked hypokalemia, or have hypertension not effectively controlled with routine antihypertensive therapy. It is important to recognize that both hypokalemia and metabolic alkalosis are no longer considered hallmarks of primary hyperaldosteronism, and in fact are absent in the majority of patients.
Diagnosing and treating primary aldosteronism is important for two major reasons. First, in many patients primary aldosteronism leads to very severe hypertension, which is poorly responsive to antihypertensive medications and thus increases cardiovascular risk. Indeed, a goal of hypertension management is to be able to identify and treat the underlying causative factor generating the individual’s hypertension. Although not feasible for the majority of patients with hypertension, this is possible for the patient with primary aldosteronism. Second, and possibly more important, untreated primary aldosteronism leads to cardiovascular events at rates significantly greater than attributable solely to the hypertension. Diagnosing and treating the primary aldosteronism, whether with adrenalectomy or with MR blockers, as clinically appropriate, corrects this increased risk.28
Pathology
Primary hyperaldosteronism can result either from an aldosterone-producing adenoma (APA) or from hyperplasia of the zona glomerulosa (Fig. 40-4). Most APAs are unilateral and are large enough (>1 cm) to be identified by computed tomography (CT) scanning (Fig. 40-5
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


