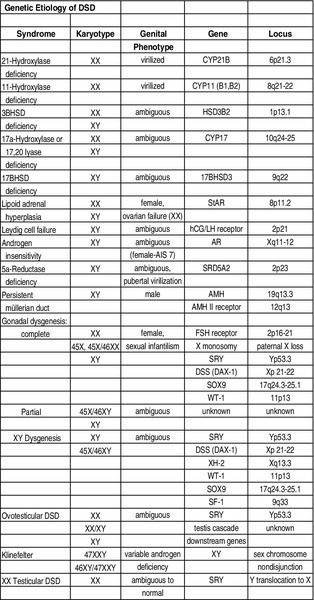
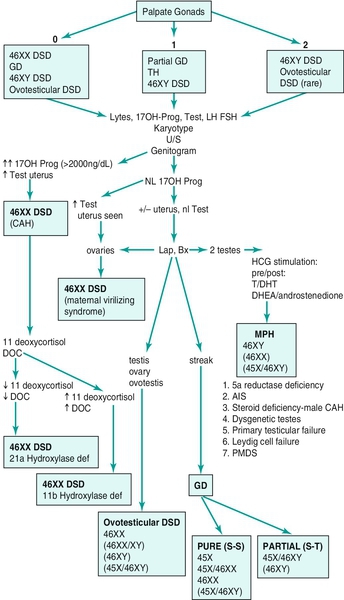
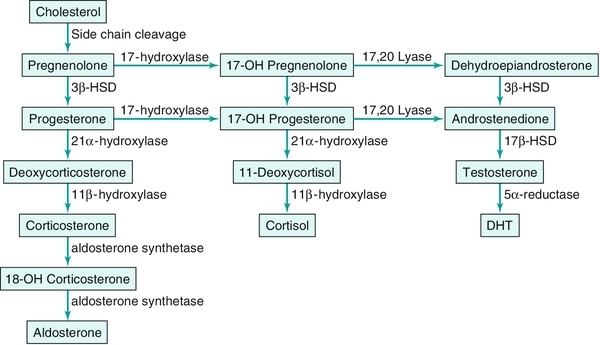
Chapter 27 Human sex development occurs in an organized, sequential manner. Chromosomal sex is established at fertilization, which then directs the undifferentiated gonads to develop into testes or ovaries. Phenotypic sex results from the differentiation of internal ducts and external genitalia under the influence of hormones and transcription factors. When discordance occurs among these three processes (i.e., chromosomal, gonadal, or phenotypic sex determination), then disorders of sex development (DSD) develop. This has previously been termed intersex or intersexualty, but the new nomenclature has ended the use of the term intersex and now refers to it as DSD. Sexual differentiation is regulated by more than 50 different genes on both the sex chromosomes and autosomes. These genes encode transcription factors, gonadal steroid and peptide hormones, and tissue receptors. The sex determination genetic cascade involves the SRY gene (sex-determining region on Y) as the testis-determining factor, although other upstream (SF1, WT1) and downstream (DAX1, SOX9, Wnt4, AMH, homeobox) genes are also likely involved (see Figure 27-e1). A failure of testis determination will usually result in the development of the female phenotype, although recent research has shed light on genes that are required in the female pathway, thus eradicating the theory that this is simply a “default” pathway. Genetic alterations resulting in partial testicular development can give rise to a wide spectrum of masculinization. In addition to defects in peptide hormones and their receptors, timing of hormonal exposure is also critical to appropriate development. Although much work remains to be done, recent advances in our knowledge have begun to unravel the molecular basis of DSD. The first sections of this chapter provide a basic template for the etiology of DSD. However, it may be worthwhile to read the history, physical exam, and evaluation descriptions first and then begin again with the start of the chapter to keep this subject in perspective. The important issue with any DSD evaluation is not to get overwhelmed by the enormity of the differential diagnosis possibilities. Instead, work slowly through the differential diagnosis pathway (see Differential Diagnosis, Page 2) based on the physical exam and laboratory findings. Many possible diagnoses will remove themselves from contention in this manner. Differential Diagnosis Decision pathway of evaluation for disorders of sex development (DSD) T = testis, S = streak gonad 46XX DSD, previously known as female pseudohermaphroditism, is the most common DSD. The ovaries and müllerian derivatives are normal and the sexual ambiguity is limited to masculinization of the external genitalia. A female fetus is masculinized if exposed to androgens, and the degree of masculinization is determined by the stage of differentiation at the time of exposure. Masculinization can occur secondary to exogenous maternal steroids as well. Congenital adrenal hyperplasia (CAH) accounts for the majority of 46XX DSD patients. Inactivating or loss of function mutations in five genes involved in steroid biosynthesis can cause CAH: CYP21, CYP11B1, CYP17, HSD3B2, and StAR (Box 27-1). Although all six of these biochemical defects are characterized by impaired cortisol secretion, only CYP21 and CYP11B1 are predominantly masculinizing disorders (HSD3B2 to a lesser extent). Although the female fetus is masculinized due to overproduction of adrenal androgens and precursors, affected males have no genital abnormalities. In contrast, HSD3B2, CYP17, and StAR deficiencies block cortisol synthesis and gonadal steroid production. Thus affected males have varying degrees of 46XY DSD (formerly male pseudohermaphroditism), whereas females generally have normal external genitalia. Each of these genetic defects is inherited in an autosomal recessive pattern. Deficiency of CYP21 (cytochrome p450c21) is the most common cause of genital ambiguity. The gene is located within the major human leukocyte antigen (HLA) locus on the short arm of chromosome 6 (6p21.3). HLA types are codominantly inherited and can be used as a marker to distinguish homozygous, heterozygous, and unaffected individuals. Two CYP21 genes are located on chromosome 6 between HLA-B and HLA-DR: a functional 10 exon CYP21B gene and a CYP21A pseudogene that are nonfunctional due to the presence of multiple stop codons. Recombination between CYP21B and the homologous but inactive CYP21A accounts for approximately 95% of 21α-hydroxylase deficiency mutations resulting in a variable decrease in 21α-hydroxylase activity. These conversions usually involve the transfer of inherent CYP21A mutations. Patients with simple virilizing 21α-hydroxylase deficiency have been noted to have a conversion mutation (Ile-Asn) causing severely decreased enzyme activity but sufficient aldosterone production to prevent salt wasting. Nonclassical CAH conversion mutations have been shown to display 20% to 50% of normal activity. Some centers have instituted screening for 17 hydroxy-progesterone levels at 3 to 5 days of life in conjunction with screening for other inborn metabolic diseases. HLA typing of amniotic fluid cells in mothers with a previously affected offspring has been used to identify a fetus with a CYP21 deficiency. This has led to experimental prenatal treatment of CAH with good results of decreasing the masculinization. At issue is the controversy of initially treating one in eight fetuses who may not need the therapy. Cytochrome p450 is a terminal oxidase found on the inner mitochondrial membrane. Cortisol deficiency results in increased secretion of 11-deoxycortisol, DOC, corticosterone, and androgen by the adrenal gland. Hypertension, which occurs in about two thirds of patients, is presumptively a consequence of excess DOC, with resultant salt and water retention. Excess androgen secretion in utero masculinizes the external genitalia of the female fetus. After birth, untreated males and females progressively masculinize and experience rapid somatic growth and skeletal maturation. Two distinct enzymes, CYP11B1 and CYP11B2, are encoded by two tandem and homologous genes at 8q21–22. CYP11B1 encodes 11β-hydroxylase, which converts 11-deoxycorticosterone (DOC) to corticosterone and 11-deoxycortisol to cortisol. This gene’s protein is expressed in the adrenal zona fasciculata and is primarily under the influence of corticotropin. Alternatively, CYP11B2 encodes for aldosterone synthetase, which converts DOC to corticosterone and 18-hydroxycorticosterone to aldosterone. It is expressed in the zona glomerulosa and is under the influence of angiotensin II and potassium. Although CYP11B1 and CYP11B2 are extremely homologous, both genes are functional. Thus gene conversions are not the cause of impaired enzyme activity, as was described for 21α-hydroxylase deficiency. Similar to CYP21, alterations with less enzyme activity usually result in more severe phenotypes, but heterogeneity can occur. This disorder can be detected prenatally, and the masculinization of the fetus can be decreased following in utero dexamethasone treatment as previously described for 21α-hydroxylase deficiency. Other abnormalities such as 3β-hydroxysteroid dehydrogenase deficiency, CYP17 deficiency, and StAR deficiency are covered under disorders of 46XY DSD. 46XY DSD is a heterogenous disorder in which testicles are present but the internal ducts and/or the external genitalia are incompletely masculinized. The phenotype ranges from completely female external genitalia to mild male ambiguity (such as hypospadias or cryptorchidism). 46XY DSD can result from eight basic etiologic categories: 2. Enzyme defects in testosterone (T) biosynthesis, some of which are common to CAH (StAR, HSD3B2, CYP17, 17-βHSD3) 3. Defects in androgen-dependent target tissues (androgen-insensitivity syndrome [AIS]) 4. A defect in the enzymatic conversion of testosterone (T) to dihydrotestosterone (DHT) (5α-reductase deficiency) 5. Defects in the synthesis, secretion, or response to antimüllerian hormone (AMH) or müllerian inhibiting substance (MIS) resulting in persistent müllerian duct syndrome 6. Aberrations in testicular gonadogenesis (testicular dysgenesis) 7. Primary testicular failure (vanishing testes) 8. Exogenous insults (maternal ingestion of progesterone/estrogen or environmental hazards) Leydig cell aplasia/hypoplasia is an autosomal recessive condition. In its complete form, these patients are 46XY but are cryptorchid and phenotypically female. Wolffian structures are present due to the secretion of MIS by intact Sertoli cells. However, the phenotypic presentation is variable. The underlying abnormality in this disorder is a failure of Leydig cell differentiation secondary to an abnormal LH receptor (LHR). LH is elevated, T is markedly decreased, and follicle-stimulating hormone (FSH) levels are unaffected. There is no T surge on hCG stimulation. Defects in four steps of the steroid biosynthetic pathway from cholesterol to T may produce genital ambiguity in the male (Figure 27-1). The StAR protein regulates the transfer of cholesterol from the outer mitochondrial membrane to the inner membrane, where it is metabolized by the CYP11A gene product, p450 scc. Congenital lipoid adrenal hyperplasia, a rare autosomal recessive disorder, was once thought to arise from mutations in the gene encoding p450 scc (cholesterol side chain cleavage), CYP11A. However, studies have shown that such mutations are incompatible with survival because these alterations would likely cause spontaneous abortions at the end of the first trimester from insufficient placental progesterone production, when corpus luteum secretion of progesterone wanes. Rather, lipoid CAH is a result of mutations in the StAR (steroidogenic acute regulatory) gene. These patients are often Asian and are born after an uneventful pregnancy. They subsequently present after a few days or weeks with severe adrenal insufficiency and will die from glucocorticoid and mineralocorticoid deficiency without hormone replacement. However, they can survive to adulthood if properly treated. 46XY males are born with female external genitalia, and the adrenal glands and testes are massively engorged with lipid deposits (cholesterol esters). Although 46XX females do experience feminization and cyclical vaginal bleeding at puberty, they invariably develop polycystic ovaries. 3β-hydroxysteroid dehydrogenase (3β-HSD) deficiency results in impaired adrenal aldosterone and cortisol synthesis and gonadal T and estradiol formation. In its complete form, severe mineralocorticoid and glucocorticoid deficiency in the first week of life are observed. Male infants exhibit variable degrees of 46XY DSD, whereas in females, clitoromegaly and mild masculinization are common. The masculinization is a result of conversion of the ∆5 precursors to T via extragonadal (placenta and peripheral tissues) fetal 3β-HSD. Salt wasting may be present, depending on the severity of the 3β-HSD deficiency. Treatment involving glucocorticoids is similar to that of 21-hydroxylase deficiency, and mineralocorticoids are added to treat the salt wasting forms. Sex steroid replacement may be necessary at puberty. The CYP17 gene is located on chromosome 10q24.3 and contains eight exons. Its protein is absent from the placenta, ovarian glomerulosa cells, and the adrenal zona glomerulosa. The cytochrome p450 c17 protein catalyzes 17α-hydroxylase and 17,20 lyase (desmolase) activities. Therefore mutations in CYP17 would be expected to alter the activities of both enzymes. Although the existence of isolated 17,20 lyase deficiency has been questioned, two patients with hormonal and clinical findings consistent with isolated 17,20 lyase deficiency have been described. Combined 17α-hydroxylase and 17,20 lyase deficiency causes deficient production of cortisol and sex hormones. Genetic females present with sexual infantilism due to a lack of ovarian estrogen synthesis at puberty, whereas genetic males often manifest a developmental spectrum ranging from a normal female phenotype to an ambiguous hypospadiac male. Overproduction of 11-DOC and corticosterone leads to hypertension, hypokalemic alkalosis, and carbohydrate intolerance. Genetic males with isolated 17,20 lyase deficiency also manifest variable degrees of incomplete masculinization. However, they do not suffer from the associated abnormalities in cortisol secretion and hypertension that occur in patients with combined 17α-hydroxylase and 17,20 lyase deficiencies. There are at least five known isoenzymes of 17βHSD. The type 3 17βHSD catalyzes the reduction of androstenedione to T and is expressed only in the testis. Different isoenzymes catalyze the isoreduction of estrone to estradiol and androstenedione to T. As expected, these patients have elevated plasma androstenedione (up to 10 times as high) levels, low plasma T (increased androstenedione-to-T ratio after hCG stimulation in prepubertal patients), elevated LH, and normal- to-high FSH levels. In these patients, more than 90% of plasma T is produced from the extragonadal conversion of androstenedione to T, compared to less than 1% in normal males. Plasma DHT may be normal in some patients, suggesting conversion from androstenedione. These patients are usually 46XY males with ambiguous or female external genitalia and normal internal wolffian structures, including inguinal testes and a blind vaginal pouch. This occurs because there is little or no peripheral conversion of androstenedione to T and DHT during early gestation by the other 17βHSD isoenzymes. Additionally, increased aromatization of androstenedione to estrogen by the placenta may leave little substrate available for conversion to T and DHT. They are often raised as girls but may virilize at puberty and adopt a male gender role, similar to that seen in 5αRD-2 deficiency. At puberty, the expression of other 17βHSD isoenzymes in the peripheral tissues partially compensates for testicular 17βHSD-3 deficiency; however, the phenotype is variable. Some patients develop gynecomastia at puberty, depending on their T-to-estradiol ratio. 5α-reductase deficiency, an autosomal recessive condition with alterations of the 5αRD-2 gene, was initially described as pseudovaginal perineoscrotal hypospadias because of the striking genital ambiguity/female external phenotype seen in these cases. These patients often have a clitorislike phallus, severely bifid scrotum, and perineoscrotal hypospadias. Some patients may have less severe genital ambiguity, however. Normal wolffian structures, cryptorchidism, and a rudimentary prostate are other common findings. At puberty, patients with 5α-reductase deficiency show signs of virilization with increased muscle mass, deepening of voice, substantial growth of phallus, rugation and hyperpigmentation of the scrotum, and normal libido. Migration of inguinal testis into the scrotum has been observed. They do not experience gynecomastia or male pattern balding, facial hair is decreased, and their prostates remain infantile. Although these patients are usually oligo- or azoospermic, fertility via intrauterine insemination has been reported. The broad phenotypic spectrum in these 46XY patients varies from normal female external genitalia (AIS7 or testicular feminization—Figures 27-2 and 27-3) to normal males with infertility (AIS1). Patients previously described by Lubs, Gilbert-Dreyfuss, Reifenstein, and others are now classified as AIS 1-7. This X-linked disorder affects 1 in 20,000 live male births. The androgen receptor (AR) gene is located in the pericentromeric region of the long arm of chromosome X at Xq11-12 and contains eight exons. The AR is a member of the nuclear receptor superfamily of transcription factors and has three major functional domains. The amino (NH2) terminal domain is encoded by exon 1 and is critical to target gene transcription regulation. Exons 2 and 3 encode the DNA-binding domain and the 5’ region of exon 4 encodes the hinge region containing the nuclear targeting signal. The 3‘ region of exon 4 and exons 5 through 8 encode the steroid-binding domain that confers ligand specificity. Binding of DHT or T to this receptor ligand-binding domain results in “activation” of the receptor. The NH2 domain contains polymorphic glutamine (CAG) and glycine (GGC) repeats (i.e., the length of these repeats varies among individuals in the general population). Hence, AR genes with different CAG and GGC repeat lengths represent different AR alleles. Interestingly, AR CAG repeat length varies by racial group: the most common Caucasian AR allele has 21 repeats, whereas the most common black allele has 18 repeats. Deletion of the CAG repeat yields an AR with increased transcriptional activity, whereas an increase in the length decreases this activity. In men with spinal bulbar muscular atrophy and Kennedy disease, the AR CAG repeat is at least twice as long as that of unaffected men. Mutations in the DNA-binding region can produce a receptor that binds androgen normally but cannot induce transactivation because of faulty binding to DNA, destabilization of the protein structure, or lack of dimerization of the receptor. The degree of impairment of transactivation correlates with the phenotype. The majority of AR gene mutations affects the steroid-binding domain and results in receptors unable to bind androgens or that bind androgens but exhibit qualitative abnormalities. Of note, however, is the finding of phenotypic variability in families with affected males with PAIS. This suggests that other factors in the sex-differentiation cascade influence the phenotypic manifestations of gene mutations.
Disorders of Sexual Development
46XX DSD (previously called female pseudohermaphroditism)
CYP21 (21A-hydroxylase) deficiency
CYP11B1 (11β-hydroxylase) deficiency
46XY DSD (previously called male pseudohermaphroditism)
Leydig cell aplasia/hypoplasia
Testosterone biosynthesis enzyme defects
StAR deficiency
3β-hydroxysteroid dehydrogenase deficiency
CYP17 abnormality
17β-hydroxysteroid dehydrogenase deficiency
5α-reductase deficiency
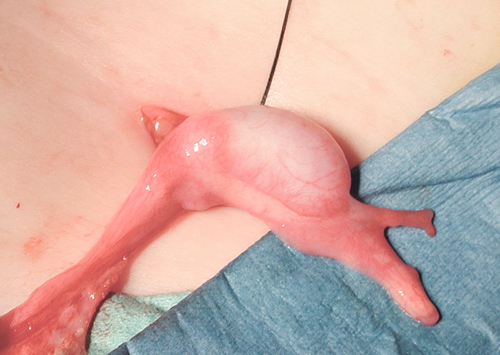
Androgen insensitivity syndrome
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


