Disorders of Potassium Balance
Potassium is the principal cation of the intracellular fluid (ICF) where its concentration is between 120 and 150 mEq/L. The extracellular fluid (ECF) and plasma potassium concentration [K] is much lower—in the 3.5–5.0 mEq/L range. The very large transcellular gradient is maintained by active K transport via the Na-K-ATPase pumps present in all cell membranes and the ionic permeability characteristics of these membranes. The resulting greater than 40-fold transmembrane [K] gradient is the principal determinant of the transcellular resting potential gradient, about −90 mV with the cell interior negative (Figure 4–1). Normal cell function requires maintenance of the ECF [K] within a relatively narrow range. This is particularly important for excitable cells such as myocytes and neurons. The pathophysiologic effects of dyskalemia on these cells result in most of the clinical manifestations.

Individual potassium intakes vary widely—a typical Western diet provides between 50 and 100 mEq K per day. Under steady-state conditions, an equal amount is excreted, mainly in urine (about 90%), and to a lesser extent in stool (5–10%) and sweat (1–10%). Normally, homeostatic mechanisms maintain plasma [K] precisely between 3.5 and 5.0 mEq/L. Rapid regulation of potassium concentration is needed to prevent potentially fatal hyperkalemia after every meal and is largely due to transcellular K shifts. The normal postprandial rise in insulin concentration moves both K and glucose into the intracellular compartment, where 98% of total body K (˜3000 mEq) is located. Postprandial insulin release is primarily related to increased plasma glucose concentrations but hyperkalemia also directly stimulates pancreatic β-cells to release insulin. Insulin deficiency and/or resistance increase plasma [K]. Epinephrine and norepinephrine also rapidly regulate transcellular K balance and become especially important during and following vigorous exercise. Hyperadrenergic states such as alcohol withdrawal and hyperthyroidism, β-sympathomimetics such as the tocolytic terbutaline, and theophylline poisoning often generate hypokalemia due to translocation of K from the ECF into cells.
Metabolic alkalosis stimulates cellular K uptake whereas some forms of hyperchloremic and other inorganic (mineral) acidoses enhance movement of K out of cells. However, the common organic metabolic acidoses (lactic and ketoacidosis) do not directly cause any K shift. Respiratory acid–base abnormalities generally have small effects. Although it had been assumed that the alkalemia produced by respiratory alkalosis would move K into cells, the opposite has been found, ie, a small increase in plasma [K] due to associated α-adrenergic stimulation. Respiratory acidosis increases plasma [K] slightly. Hyperosmotic conditions that shift fluid out of cells are an important cause of K translocation to the ECF. Finally, hypokalemia per se moves K from the intracellular to the extracellular space.
Potassium absorption in the small intestine is not specifically controlled. Although colonic epithelial cells can increase K secretion in response to chronic hyperkalemia (patients with chronic kidney disease), the net effect on K balance is minor. Although the [K] in stool water may be high, the quantity of water in formed stool is small—thus absent diarrhea, total stool K excretion is low and most ingested K is absorbed.
Potassium excretion is principally into the urine and the main regulator of body K balance is the kidney. Potassium is freely filtered (600–800 mEq/day) and then largely reabsorbed in the proximal tubule and thick ascending loop of Henle. The K load delivered to the cortical collecting duct (CCD) is about 10–15% of filtered K and the intraluminal [K] of fluid entering this segment is low. It is here that major regulation of K excretion occurs. Sodium (Na) reabsorption and K and H secretion in the CCD is dependent on the amount of Na delivered to this segment, the “absorbability” of the anion, and the activity of the mineralocorticoid aldosterone. In the CCD, Na is absorbed through epithelial Na channels (ENaC) present on the luminal surface of the predominant (principal) cells in this segment. The absorption of large amounts of Na, especially when delivered with an anion not easily absorbed (Cl, HCO3, and others), generates a negative charge within the lumen and enhances the secretion of K and H (Figure 4–2). Aldosterone regulates the rate of Na absorption through these channels at multiple levels. It affects the rate of energy (ATP) generation, the activity of Na-K-ATPase pumps, and the number and “open state” of the ENaC channels themselves.
Figure 4–2.
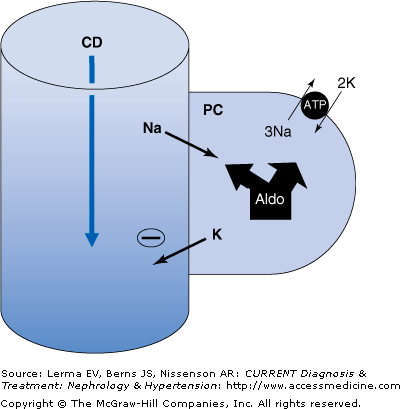
K handling by the cortical collecting duct. Aldosterone has multiple effects on electrolyte transport in the cortical collecting duct (CCD). Sodium (Na) absorption increases through stimulation of basolateral Na-K-ATPase activity and the increased number and “open state” of the luminal Na channel (ENaC). The influx of Na causes a negative charge to develop within the lumen. This stimulates K (and H) secretion into the lumen down electrical and chemical gradients. Volume-contracted states result in little Na delivery to the CCD (due to avid more proximal absorption) so that K (and H) secretion is slight despite high aldosterone levels. Volume-expanded states enhance delivery of Na to the CCD and cause physiologically adequate levels of K (and H) secretion due to suppressed aldosterone levels.
In normal individuals, an inverse relationship exists between aldosterone activity and CCD Na delivery. A high salt intake will expand ECF volume, inhibit renin and aldosterone levels, and increase distal delivery and excretion of Na. High distal Na delivery counterbalances low aldosterone activity and the net effect is normal CCD K and H secretion and excretion. Conversely, a low salt intake contracts the ECF, stimulates renin and aldosterone levels, and markedly reduces distal CCD Na delivery and excretion. In this case, low CCD Na delivery is linked to high aldosterone activity and again normal K and H secretion and excretion is maintained. This reciprocal interplay between aldosterone activity and distal Na delivery is physiologic and acts to simultaneously maintain both volume and electrolyte homeostasis.
Pathophysiologic conditions exist when high CCD Na delivery combines with high aldosterone activity or low CCD Na delivery coexists with low aldosterone activity. In the first circumstance, the magnitude of CCD Na absorption increases markedly and results in excessive K and H secretion, thereby generating hypokalemia and metabolic alkalosis. Conversely, the second scenario causes a major reduction in CCD Na reabsorption and very low rates of K and H secretion leading to hyperkalemia and metabolic acidosis. Increased CCD Na delivery together with high aldosterone levels occurs in patients with primary hyperaldosteronism and as a result of thiazide and/or loop diuretics. Reduced CCD Na delivery together with low aldosterone activity occurs in some forms of hyporeninemic hypoaldosteronism and when aldosterone antagonists are given to patients with disorders with reduced “effective” intra-arterial volume such as hepatic cirrhosis or congestive heart failure.
Nerve, cardiac conduction, and muscle cells are especially sensitive to changes in transcellular voltage and therefore are most affected by hypokalemia or hyperkalemia. Figure 4–3 shows how either condition can cause muscle weakness.
Figure 4–3.
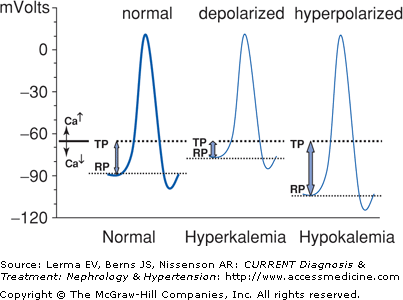
Cell depolarization and hyperpolarization depend on extracellular potassium. An action potential is generated when the cell depolarizes from its resting potential (RP) to the threshold potential (TP). Hyperkalemia moves the RP closer to the TP and results in depolarization muscle paralysis. Hypokalemia hyperpolarizes the cell and thereby impairs depolarization. The flaccid paralysis caused by hypokalemia or hyperkalemia is clinically similar. Calcium raises the TP, ameliorating the effects of hyperkalemia, while hypocalcemia has the opposite effect.
Hypokalemia increases the resting potential across the myocyte membrane, ie, the cell becomes more negative and less sensitive to excitation. Severe hypokalemia thus leads to a hyperpolarization block and flaccid paralysis. It may also cause rhabdomyolysis and paralytic ileus. Renal manifestations include metabolic alkalosis, nephrogenic diabetes insipidus, and formation of renal cysts. Chronic hypokalemia has been implicated in the development of hypertension.
Hyperkalemia reduces the resting potential, ie, the cell becomes less negative. Following depolarization, the cell is unable to adequately repolarize and becomes unexcitable. Severe hyperkalemia causes a depolarization block and flaccid paralysis. Clinical manifestations include fatigue, myalgia, and muscle weakness (especially lower extremity), hyporeflexia, paresthesias, muscle cramps, electrocardiogram (ECG) changes, and cardiac arrhythmia (Figure 4–4). Muscle weakness may progress to ascending paralysis, hypoventilation, and respiratory failure.
Figure 4–4.
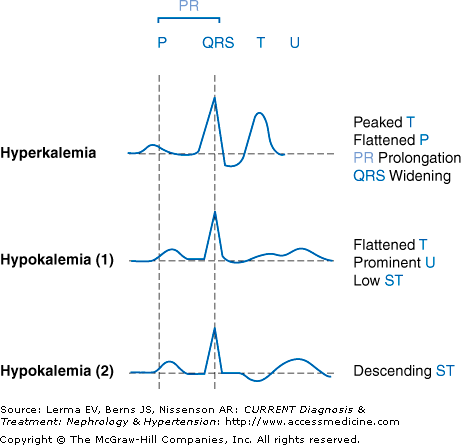
Electrocardiographic tracings with hypokalemia and hyperkalemia. Hyperkalemia initially causes peaking (“tenting”) of T waves and then progresses to widening of the QRS and PR intervals, sinus bradycardia and arrest, atrioventricular (AV) block, fusion of QRS with T (sine wave appearance), idioventricular rhythm, and finally ventricular tachycardia and fibrillation, and asystole. Hypokalemia causes ST depression, flattening of the T waves, and prominent U waves. This progresses to fusion of the T and U waves into a single wave and the ST segment becomes negative and descending. The QT interval lengthens, especially if hypocalcemia or hypomagnesemia is present. Atrial and ventricular arrhythmias may develop.
The clinical manifestations of an abnormal plasma [K] vary greatly and depend on (1) its magnitude, (2) acuity of onset, (3) the relative contributions of K shift versus change in total body K, and (4) coexisting abnormalities that either potentiate or blunt the [K] effects, including underlying heart disease, drugs (digoxin, antiarrhythmic agents), hypocalcemia or hypercalcemia, cardiac pacing devices, and others.
The resting membrane potential is determined by the ratio of intracellular and extracellular [K] (Ki/Ke). An acute K shift into or from the intracellular space alters intracellular [K] only minimally since it is quantitatively so large; about 3000 mEq or 98% of total body K is within cells. However, the effect on the extracellular concentration can be dramatic because the total quantity of K outside of cells is only about 60 mEq. Therefore, acute K shifts will markedly affect the Ki/Ke ratio and can cause profound cellular hyperpolarization or depolarization with muscular, neurologic, and cardiac symptoms (Figure 4–5). In contrast, states of chronic K depletion reduce both intracellular and extracellular K levels and have a much smaller effect on Ki/Ke with fewer and less severe clinical manifestations. Furthermore, K shifts produce much more rapid changes in plasma [K] and thus the effects are often more dramatic than with states of total body K depletion or excess. This has important clinical implications: A dialysis patient with a chronically elevated [K] who presents with a [K] of 6.5 mEq/L and minimal ECG effects may not require urgent intervention. However, a diabetic patient with chronic kidney disease who develops acute hyperglycemia and a rapid [K] rise to 6.5 mEq/L may manifest major ECG changes, which mandate quick action.
Figure 4–5.
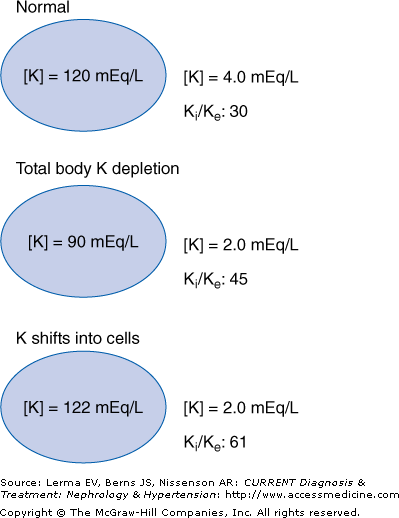
K distribution with intracellular K shift versus K depletion. The resting membrane potential is determined by the ratio of intracellular and extracellular potassium (Ki/Ke). Total body K depletion reduces both intracellular and extracellular [K]. The Ki/Ke ratio increases and the cell becomes hyperpolarized. A transcellular shift of K into cells slightly increases intracellular [K] and markedly reduces extracellular [K]. Therefore, the Ki/Ke ratio increases markedly and cellular hyperpolarization is severe and often produces clinical symptoms.
Comorbid illness such as coronary heart disease will amplify the clinical importance of dyskalemia by increasing the risk of serious arrhythmia. Hyperkalemic effects on cardiac conduction are well documented and are the principal reason it constitutes a medical emergency (Figure 4–4). The cardiac risks of hypokalemia are less well established. Although increased risk of ectopy is established for patients with acute myocardial infarction and those treated with digoxin and other antiarrhythmic agents, its importance for most other patients remains unclear.
Calcium has important effects on myocyte depolarization. Hypocalcemia reduces the depolarization threshold potential and renders the cardiac myocyte more excitable. Conversely, hypercalcemia reduces membrane excitability by increasing the depolarization threshold (Figure 4–3). This calciumrelated shift of the depolarization threshold reverses the cardiac toxicity of hyperkalemia. Coexisting hyperkalemia and hypocalcemia is a particularly pernicious combination and is common in patients with severe kidney failure.
Hypokalemia
- Serum [K] below 3.5 mEq/L.
- Most commonly caused by use of thiazide or loop diuretics, vomiting/nasogastric suction, and diarrhea (or laxatives).
- Reduction of total body K stores.
- Excretion less than 20–30 mEq K per day.
- Osmotic diuresis.
A serum [K] below 3.5 mEq/L defines hypokalemia, and Table 4–1 lists some of the causes and clinical conditions associated with this disorder. The most common causes of hypokalemia are the use of thiazide or loop diuretics, vomiting/nasogastric suction, and diarrhea (or laxatives). These etiologies are usually readily apparent unless the patient is covertly using drugs or vomiting. A more elaborate evaluation is necessary when these frequent causes are excluded. It is then important to determine whether hypokalemia is primarily due to an intracellular shift of K or to excessive K renal or gastrointestinal losses (sometimes combined with reduced intake).
Renal losses |
Diuretics |
Vomiting, nasogastric suction |
Osmotic diuresis (uncontrolled diabetes and others) |
Drugs |
Excretion of nonreabsorbable anions |
Fludrocortisone and others |
Licorice (see text) |
Tubular toxicity (aminoglycosides, cisplatinum, and others) |
Primary hyperaldosteronism |
Liddle syndrome |
Cushing’s disease |









