Disorders of Phosphate Balance: Hypophosphatemia & Hyperphosphatemia: Introduction
New data demonstrate that serum phosphate, similar to serum calcium, is a signaling molecule. The mechanisms for sensing serum phosphate signal transduction concentration are not understood. This chapter considers serum phosphorus in a context greater than mineral and metabolic homeostasis. The bulk of total body phosphate (85%) is in the bone as part of the mineralized extracellular matrix. This phosphate pool is accessible, albeit in a limited fashion through bone resorption. Phosphate is a predominantly intracellular anion with an estimated concentration of approximately 100 mmol/L, most of which is either complexed or bound to proteins or lipids. Serum phosphorus concentration varies with age, time of day, fasting state, and season. It is higher in children than adults. Phosphorus levels exhibit a diurnal variation with the lowest phosphate level occurring near noon. Serum phosphorus concentration is regulated by diet, hormones, and physical factors such as pH. Importantly, because phosphate moves in and out of cells under several influences, the serum concentration of phosphorus may not reflect phosphate stores.
Hyperphosphatemia
- Serum inorganic phosphorus (Pi) concentration greater than 2.5–4.5 mg/dL or 0.75–1.45 mM in adults or 6 or 7 mg/dL in children.
- Consequence of increased intake of Pi, decreased renal excretion of Pi, or translocation of Pi from tissue breakdown into extracellular fluid.
- Short-term consequences are hypocalcemia and tetany.
- Long-term consequences are soft tissue calcification and secondary hyperparathyroidism.
Serum inorganic phosphorus (Pi) concentrations are generally maintained at 2.5–4.5 mg/dL or 0.75–1.45 mM in adults, whereas hyperphosphatemia is not present in children unless serum Pi levels are greater than 6 or 7 mg/dL. Hyperphosphatemia may be the consequence of an increased intake of Pi, a decreased excretion of Pi, or translocation of Pi from tissue breakdown into the extracellular fluid (Table 7–1). Because the kidneys are able to excrete phosphate very efficiently over a wide range of dietary intake, hyperphosphatemia most frequently results from renal insufficiency and the attendant inability to excrete Pi. However, in metabolic bone disorders such as osteoporosis and renal osteodystrophy, the skeleton is a poorly recognized contributor to serum phosphorus.
Increased intake |
Intravenous—sodium or potassium phosphate |
Oral administration—Neutraphos |
Rectal—Fleets phosphosoda enemas |
Decreased renal excretion |
Renal insufficiency/failure—acute or chronic |
Pseudohypoparathyroidism |
Tumoral calcinosis |
Hypoparathyroidism |
Acromegaly |
Bisphosphonates |
Childhood |
Excess bone resorption |
Transcellular shift from intracellular to extracellular spaces |
Catabolic states |
Fulminant hepatitis |
Hyperthermia |
Rhabdomyolysis—crush injuries or nontraumatic |
Cytotoxic therapy—tumor lysis |
Acute leukemia |
Diabetic ketoacidosis |
Hemolytic anemia |
Acidosis—metabolic or respiratory |
Artifactual |
Hyperphosphatemia can be the consequence of an increased intake or administration of Pi. Intravenous administration of Pi during parenteral nutrition, the treatment of Pi depletion, or hypercalcemia can cause hyperphosphatemia, especially in patients with underlying renal insufficiency. Hyperphosphatemia may also result from overzealous use of oral phosphates or of phosphate-containing enemas as phosphate can be absorbed passively from the colon through paracellular pathways. Administration of vitamin D and its metabolites in pharmacologic doses may be responsible for the development of hyperphosphatemia, although suppression of parathyroid hormone (PTH) and hypercalcemia-induced renal failure are important pathogenetic factors in this setting.
Clinically, hyperphosphatemia is most often caused by impaired excretion due to kidney failure. During stages II and III of chronic kidney disease (CKD), phosphate balance is maintained by a progressive reduction in the fraction of filtered phosphate reabsorbed by the tubules leading to increased Pi excretion by the remaining nephrons and a maintenance of normal renal Pi clearance. In advanced kidney failure, the fractional excretion of Pi may be as high as 60–90% of the filtered load of phosphate. However, when the number of functional nephrons becomes too diminished (glomerular filtration rate usually < 20 mL/minute) and dietary intake is constant, Pi balance can no longer be maintained by reductions of tubular reabsorption, and hyperphosphatemia develops. When this occurs, the filtered load of Pi per nephron increases and Pi excretion rises. As a result, Pi balance and renal excretory rate are reestablished, but at a higher serum Pi level (hyperphosphatemia). An unresolved issue during late stages of CKD, stage IV with persistent hyperphosphatemia and elevated PTH levels, there is phosphate reabsorption. The fractional excretion may be 80%, but why isn’t it 95% and balance maintained with normophosphatemia?
Defects in renal excretion of Pi in the absence of renal failure may be primary, as in pseudohypoparathyroidism (PHP). PHP is a term for a group of disorders characterized by hypocalcemia and hyperphosphatemia due to resistance to the renal tubular actions of parathyroid hormone. As a result of the molecular abnormalities in PHP, PTH does not decrease proximal renal tubular phosphate transport causing hyperphosphatemia.
A second primary defect in renal Pi excretion is tumoral calcinosis. This is usually seen in young black males with ectopic calcification around large joints and is characterized by increased tubular reabsorption of calcium and Pi and normal responses to PTH. Familial forms of tumoral calcinosis are due to mutations in the UDP-N-acetyl-α-d-galactosamine: polypeptide N-acetylgalactosaminyltransferase 3 (GALNT3) gene and missense mutations in fibroblast growth factor (FGF)23. It has been demonstrated that besides PTH, FGF23 is a second hormonal regulator of renal phosphate transport in physiologic conditions.
Secondary tubular defects in phosphate transport include hypoparathyroidism and high blood levels of growth hormone in acromegaly. Finally, bisphosphonates such as Didronel (disodium etidronate), Pamidronate, or Alendronate may cause hyperphosphatemia. The mechanisms of action are unclear, but they may involve cellular phosphate redistribution and decreased renal excretion. Serum phosphorus values are normally elevated in children as compared to adults.
Clinical and translational studies demonstrate that excess bone resorption contributes to the level of serum phosphorus, which is poorly appreciated. Even in clinical situations where bone formation is decreased (adynamic) such as osteoporosis, a variability in the serum phosphorus is produced when bone formation is stimulated (Figure 7–1). For instance, in low turnover osteodystrophy treated with a skeletal anabolic agent, if phosphorus intake is constant the serum phosphorus falls despite no change in or a decrease in phosphate excretion (Figure 7–1B).
Figure 7–1.

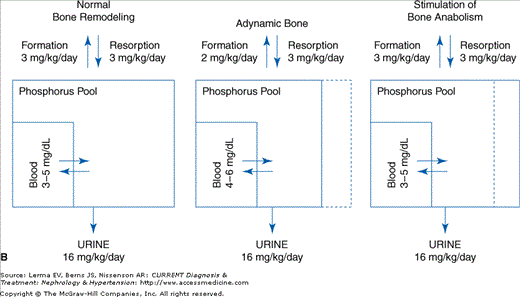
Skeletal remodeling contributes to phosphate balance and serum phosphorus levels. A: The phosphate balance diagram is amplified to show that the serum phosphorus is a small component of a rapidly exchangeable phosphorus pool composed of cellular phosphorus and the bone mineralization front. B: When bone formation is decreased (adynamic bone disorders) the exchangeable pool size is diminished and intestinal absorption from food intake will produce larger fluctuations in the serum phosphorus. These fluctuations are sufficient to activate the signaling actions of the serum Pi even though the fasting serum Pi is normal. Stimulation of bone anabolism increases the exchangeable phosphorus pool size and decreases serum phosphorus fluctuations. In end-stage kidney disease, treatment of secondary hyperparathyroidism with a calcimimetic that does not affect phosphate absorption decreases serum phosphate demonstrating the role of the skeleton in hyperphosphatemia.
Transcellular shift of Pi from cells into the extracellular fluid compartment may lead to hyperphosphatemia, as seen in conditions associated with increased catabolism or tissue destruction (eg, systemic infections, fulminant hepatitis, severe hyperthermia, crush injuries, nontraumatic rhabdomyolysis, and cytotoxic therapy for hematologic malignancies such as acute lymphoblastic leukemia and Burkitt’s lymphoma). In the “tumor lysis syndrome,” serum Pi levels typically rise, due to release from dying cells, within 12 days after initiation of treatment. The rising serum Pi concentration is often accompanied by hypocalcemia, hyperuricemia, hyperkalemia, and renal failure. Patients with diabetic ketoacidosis commonly have hyperphosphatemia at the time of presentation despite total body Pi depletion. Insulin, fluid, and acid-base therapy are accompanied by a shift of Pi back into cells and the development of hypophosphatemia. In lactic acidosis, hyperphosphatemia likely results from tissue hypoxia with a breakdown of adenosine triphosphate (ATP) to adenosine monophosphate (AMP) and Pi.
Hyperphosphatemia may be artifactual when hemolysis occurs during the collection, storage, or processing of blood samples.
The most important short-term consequences of hyperphosphatemia are hypocalcemia and tetany, which occur most commonly in patients with an increased Pi load from any source, exogenous or endogenous. By contrast, soft tissue calcification and secondary hyperparathyroidism are long-term consequences of hyperphosphatemia that occur mainly in patients with renal insufficiency and decreased renal Pi excretion.
With rapid elevations of serum Pi, hypocalcemia and tetany may occur with serum Pi concentrations as low as 6 mg/dL, a level that if reached more slowly has no detectable effect on serum calcium. Hyperphosphatemia, in addition to its effect on the calcium × phosphate ion product with resultant calcium deposition in soft tissues, also inhibits the activity of 1α-hydroxylase in the kidney, resulting in a lower circulating level of 1,25-dihydroxyvitamin D3 . This further aggravates hypocalcemia by impairing intestinal absorption of calcium and inducing a state of skeletal resistance to the action of PTH.
Phosphate-induced hypocalcemia is common in patients with acute or chronic renal failure, and usually develops slowly. Tetany is uncommon unless a superimposed acid–base disorder produces an abrupt rise in plasma pH that acutely lowers the serum ionized calcium concentration. Profound hypocalcemia and tetany are occasionally observed during the early phase of the “tumor lysis” syndrome and rhabdomyolysis.
Extraskeletal calcification associated with hyperphosphatemia is usually seen in patients with CKD, diabetes, severe atherosclerosis, and aging. Recent basic, translational, and clinical research studies have led to new theories concerning the pathogenesis and the consequences of this phenomenon. Several inhibitors of vascular calcification have been discovered, including osteoprotegerin, osteopontin, matrix GLA protein, the klotho gene product, and Smad6. These substances constitute an inherent defense against heterotopic mineralization, which is breached in the disease environment. In the setting of CKD, hyperphosphatemia has been identified as a major factor contributing to the forces favoring mineralization.
In contrast to the breach of defense theory of vascular calcification, there is significant evidence that vascular cells undergo osteogenesis resulting in vascular mineralization. Experimental models have demonstrated that elevated phosphate is a direct stimulus of this transformation. The finding of vascular calcification and the role of hyperphosphatemia have more than academic significance. Calcification of the neointima or the tunica media including the large blood vessels, coronary arteries, and heart valves in patients with renal failure and patients with diabetes is associated with a high morbidity and mortality from systolic hypertension, congestive heart failure, coronary artery disease, and myocardial infarction. Another manifestation of vascular calcification in more peripheral arteries, calciphylaxis, is also associated with hyperphosphatemia and carries a poor prognosis. As a result, both vascular calcification and hyperphosphatemia are independent risk factors for cardiovascular disease and mortality.
Occasionally, an acute rise in serum Pi (eg, during Pi treatment for hypercalcemia or vitamin D intoxication) may lead to soft tissue calcification in clinical settings in addition to those previously mentioned. The blood vessels, skin, cornea (band keratopathy), and periarticular tissues are common sites of calcium phosphate deposition.
Hyperphosphatemia due to renal failure also plays a critical role in the development of secondary hyperparathyroidism and renal osteodystrophy and in mortality. Several mechanisms contribute to these complications including hyperphosphatemia-induced hypocalcemia through physicochemical interactions, expression of tumor growth factor (TGF)-α and the epidermal growth factor receptor (EGFR) in parathyroid chief cells leading to hyperplasia and increased PTH secretion and inhibition of vitamin D synthesis, and hyperphosphatemia-stimulated vascular calcification. In patients with advanced renal failure, the enhanced phosphate load from PTH-mediated osteolysis may ultimately become the dominant influence on serum phosphorus levels (Figure 7–1B). This phenomenon may account for the correlation between serum phosphorus levels and the severity of osteitis fibrosa cystica in patients maintained on chronic hemodialysis. Hyperphosphatemia also plays a critical role in the development of vascular calcification as previously discussed. There is a direct relationship between defective orthotopic mineralization (bone formation) in CKD and increased heterotopic mineralization. Data demonstrate that increasing bone formation will lower phosphate levels and diminish vascular calcification in CKD.
Finally, it should be noted that the clinical and translational data derived from the study of CKD in soft tissue calcification indicate that phosphorus is a heretofore unrecognized risk factor for cardiovascular disease.
Correction of the pathogenetic defect should be the primary aim in the treatment of hyperphosphatemia. When hyperphosphatemia is due solely to increased intake, discontinuation of supplemental phosphate and maintenance of adequate volume for diuresis are generally sufficient, as the kidneys will promptly excrete the excess. In the uncommon circumstance of significant hyperphosphatemia due to transcellular shift, treatment should be dictated by the underlying cause. For example, hyperphosphatemia that accompanies diabetic ketoacidosis will resolve with insulin therapy, as insulin stimulates cellular uptake of phosphate. On the other hand, hyperphosphatemia seen with tumor lysis, rhabdomyolysis, or other conditions characterized by massive cell death or injury should be treated as an excess phosphate load, albeit endogenous instead of exogenous. Limitation of phosphate intake and enhanced diuresis will generally resolve this cause of hyperphosphatemia, provided renal function is adequate.
When renal insufficiency is present, however, the most effective way to treat hyperphosphatemia is to reduce dietary Pi intake and to add phosphate-binding agents. Because Pi is present in almost all foodstuffs, rigid dietary phosphate restriction requires a barely palatable diet that few patients can accept. However, dietary Pi can be reduced to 800–1000 mg/day with modest protein restriction. A predialysis level of 4.5–5.0 mg/dL is reasonable and allows some room for removal of phosphorus with dialysis while avoiding severe postdialysis hypophosphatemia. To achieve this, the addition of phosphate binders to reduce intestinal absorption of dietary Pi is required. Calcium salts and sevelamer have replaced aluminum salts as first-line Pi binders. However, calcium salts contribute to the calcium phosphate ion product, and massive calcium intake is often required to maintain serum phosphorus in the target range.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


