Disorders of Magnesium Balance
Magnesium is the second most abundant intracellular cation and the fourth most common cation in the human body. It plays an essential role in a variety of cellular processes including enzyme activities involving adenosine triphosphate (ATP), energy metabolism, nucleic acid and protein synthesis, regulation of ion channels, and stabilization of membrane structures. The importance of magnesium in the body is reflected in the diverse clinical effects that accompany disorders of magnesium homeostasis. The average size adult contains approximately 24 g (1 mol, 2000 mEq) of magnesium. It is predominantly stored in bone (55–60%) and the intracellular compartments of muscle (20%) and soft tissues (20%) and it exchanges very slowly with extracellular magnesium. Therefore, skeletal and intracellular magnesium is an ineffective buffer in the setting of acute extracellular magnesium loss.
Approximately 1% of total body magnesium is in the extracellular fluid (ECF) and is composed of three fractions: 60–65% is free, ionized, and physiologically active; 30% is protein bound; and the balance is complexed to citrate, phosphate, and other anions. In clinical practice, magnesium status is assessed by measurement of total serum magnesium. Serum magnesium concentrations normally average 1.7–2.3 mg/dL (1.4–2.1 mEq/L). Given the intracellular nature of this cation, serum magnesium concentrations poorly reflect total body status.
Daily magnesium intake in the typical American diet averages 300–360 mg/day. Food sources of magnesium include green vegetables, nuts, and whole grains, as well as some meats and seafood. Of dietary magnesium 30–40% is absorbed in the gut, primarily by the small intestine, with smaller amounts being absorbed in the colon. There is some magnesium in intestinal secretions (approximately 20–40 mg), but under normal circumstances their contribution to overall magnesium elimination is minimal. However, these losses can become quite substantial in diarrheal states or with biliary fistulas.
The kidney is the main organ responsible for magnesium homeostasis. Approximately 70–80% (2.4 g) of the total serum magnesium is filtered by the kidneys. Under normal circumstances 95–97% is reabsorbed by the tubules. The plasma magnesium concentration is the most important determinant of renal magnesium excretion. Less than 5% (120 mg) is normally excreted in urine. However, hypomagnesemia results in conservation of magnesium by normal kidneys leading to a fractional excretion of less than 0.5% (12 mg) per day. Conversely, the kidneys increase excretion of magnesium to approximate the filtered load during periods of increased intake or excess magnesium administration.
In contrast to many of the other electrolytes (ie, Na+, K+, and Ca2+), control of magnesium reabsorption does not appear to be tightly regulated by a specific hormone. Parathyroid hormone, calcitonin, vitamin D, glucagon, antidiuretic hormone, aldosterone, sex steroids, and β-adrenergic agonists can affect magnesium handling in experimental studies, but it is not known if these effects have an important role in humans.
While the proximal tubule is the major site of reabsorption of other ions, only a small percentage (15–25%) of the filterable magnesium is reabsorbed in this segment. Here, magnesium transport is passive, driven by bulk flow, and depends on sodium reabsorption. Factors that affect sodium reabsorption (ie, volume expansion) can also affect magnesium reabsorption. The majority of magnesium reabsorption (60–70%) occurs in the cortical thick ascending limb (TAL) of the loop of Henle. Here again, magnesium reabsorption is a passive, paracellular process and depends on sodium reabsorption. The driving force for the reabsorption of magnesium (and calcium) is the lumen-positive electrical potential generated by sodium chloride reabsorption via the Na+/K+/2Cl− cotransporter in concert with the coordinated activity of the basolateral Na+ − K+-ATPase, a chloride channel, and an apical membrane potassium channel. Disturbances of this coordinated activity at any site (such as with loop diuretics or inherited defects/Bartter’s syndrome) will abolish the lumen-positive gradient needed to drive magnesium reabsorption and, thus, result in magnesium wasting.
The potassium channel can be secondarily inhibited by the activation of the Ca2+/Mg2+ sensing receptor (CaSR). The CaSR binds both magnesium and calcium. This accounts for the observed magnesium wasting seen in the setting of hypercalcemia that augments activation of this receptor. If the positive transepithelial gradient is ultimately generated, the paracellular reabsorption of magnesium (and calcium) occurs passively, facilitated by the tight junction protein, paracellin-1 (claudin-16). The fact that both calcium and magnesium travel in parallel through the same channel in this part of the nephron explains why disturbances resulting in hypermagnesuria will simultaneously cause hypercalciuria. The mechanism(s) of magnesium reabsorption in the distal convoluted tubule (DCT) is less well understood. Although the distal nephron accounts for only approximately 5–10% of magnesium reabsorption, it does play an important role in determining the final urinary concentration of magnesium. Reabsorption is active and transcellular and is probably mediated by Mg2+ selective channels and a basolateral membrane Na+/Mg2+ exchanger.
Hypomagnesemia
- Serum magnesium level, <1.5 mg/dL.
- A normal serum magnesium level does not exclude the diagnosis of total body magnesium depletion.
- Hypomagnesemia is a relatively common disorder, occurring in 12% of hospitalized patients and in up to 60–65% of Intensive Care Unit (ICU) patients.
- Evidence suggests that the presence of hypomagnesemia in the ICU patient population is associated with increased morbidity and mortality.
- There are conflicting data regarding the benefits of preventing hypomagnesemia, possible preventive treatment strategies, and even the level of hypomagnesemia that should prompt supplementation.
The diversity of the cellular processes in which magnesium has been shown to take part is reflected by the diversity of symptoms attributed to magnesium deficiency (Table 8–1). Hypomagnesemia may be asymptomatic, particularly if it is mild and if it develops slowly. Severe hypomagnesemia, particularly if it develops rapidly, can be associated with signs and symptoms related to cardiovascular, neuromuscular, and central nervous system (CNS) dysfunction.
General | Apathy, depression, confusion, anorexia |
|---|---|
Cardiovascular | Cardiac arrhythmias (torsades de pointes, ventricular and supraventricular) |
Increased digitalis sensitivity | |
EKG changes: widening of QRS, prolonged PR/QR intervals, T wave changes | |
Neuromuscular | Chvostek’s and Trousseau’s signs |
Muscle fasciculations and cramps | |
Tetany | |
Seizures | |
Muscle weakness | |
Obtundation | |
Electrolyte abnormalities | Hypokalemia |
Hypocalcemia |
Magnesium regulates several cardiac ion channels including the calcium channel and outward potassium currents. Lowering myocardial cytosolic magnesium can lead to shortening of the action potential and an increased susceptibility to tachyarrhythmias, particularly of ventricular origin (including torsades de pointe, monomorphic ventricular tachycardia, and ventricular fibrillation). This is particularly true in acutely ill patients and in the setting of acute myocardial infarction, congestive heart failure, or after cardiopulmonary bypass surgery. Hypomagnesemia can magnify digitalis cardiotoxicity as both the cardiac glycoside and magnesium depletion reduce intracellular potassium by inhibition of the Na+-K+-ATPase. The EKG changes associated with hypomagnesemia include progressive widening of the QRS complex, prolongation of the PR interval, and abnormalities of T wave morphology.
Hypomagnesemia augments skeletal muscle contraction and delays muscle relaxation. Therefore, affected patients can develop signs of neuromuscular irritability including tremor, muscle twitching, Trousseau and Chvostek signs, and frank tetany. These signs may be exacerbated by a coexistent electrolyte abnormality such as hypocalcemia. Patients may also present with delirium, coma, or seizures.
Electrolyte disturbances associated with symptomatic magnesium depletion include hypokalemia and hypocalcemia, both of which can be refractory to treatment unless the underlying magnesium deficit is corrected. The hypokalemia that frequently accompanies hypomagnesemia may be due to (1) a direct effect of hypomagnesemia on potassium channels in the loop of Henle (and perhaps the cortical collecting tubule) due to impairment of the Mg-dependent Na+-K+-ATPase leading to renal potassium wasting; and (2) the underlying disorders (ie, diarrhea, diuretics) that simultaneously cause magnesium and potassium loss. The hypocalcemia that often accompanies severe magnesium depletion is due to the suppressive effect of hypomagnesemia on parathyroid secretion as well as skeletal resistance to parathyroid hormone (PTH). In addition, low plasma levels of calcitriol (1, 25-dihydroxyvitamin D) have been noted in hypomagnesemic states and can contribute to the fall in calcium concentrations.
Normomagnesemic magnesium depletion (total body magnesium depletion in normomagnesemic patients) should be considered in patients at risk for magnesium depletion who have clinical features consistent with magnesium depletion, such as unexplained hypocalcemia or hypokalemia.
The terms hypomagnesemia and magnesium deficiency tend to be used interchangeably. However, because only a small fraction of magnesium is extracellular, the serum magnesium level is not a reliable way to assess total body magnesium depletion. The total body may be markedly depleted before the serum level drops. Hence, a normal magnesium level does not rule out the possibility of a magnesium deficit. Clues to the diagnosis of true magnesium depletion despite normal measured levels include persistent, unexplained hypocalcemia or hypokalemia, which is refractory to treatment or response to empiric treatment. The magnesium retention test, which measures urinary excretion of magnesium in response to an intravenous magnesium load, has also been used to assess total body magnesium status in patients suspected of having hypomagnesemia. When magnesium stores are deficient, more of the infused magnesium will be reabsorbed and, thus, less will be excreted in the urine. If less than 50% of the infused magnesium is recovered in the urine, magnesium deficiency is likely. However, this test is not in routine use as its utility is questionable and several conditions (ie, impaired renal function and renal magnesium wasting) and drugs can lead to invalid results.
If laboratory tests confirm hypomagnesemia, the next step is to distinguish between renal and extrarenal (gastrointestinal or miscellaneous) causes of magnesium wasting. A review of the clinical history can often provide this information (ie, chronic diarrhea causing excessive gastrointestinal magnesium losses). If the cause is not readily apparent, quantitative assessment of urinary magnesium excretion with a 24-hour urine collection or the calculation of the fractional excretion of magnesium (FeMg) on a random urine specimen can provide insight. In the setting of magnesium depletion, conservation of magnesium by normal kidneys can decrease the usual fractional excretion of magnesium from 3% (approximately 100 mg) to very low levels (ie, sometimes less than 0.5% or 12 mg/day). Therefore, demonstrating an inappropriately high rate of renal magnesium excretion in the setting of hypomagnesemia confirms the diagnosis of renal magnesium wasting. Table 8–2 summarizes the urine tests and the criteria used for renal magnesium wasting.
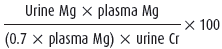
There are multiple causes of hypomagnesemia (Table 8–3). When the cause is not obvious from the clinical history and examination, it is often helpful for the clinician to try to ascertain whether the cause is due to redistribution of extracellular magnesium into the intracellular compartment, a gastrointestinal source, urinary magnesium wasting, or “complex causes.”
High urine magnesium | Low urine magnesium |
|---|---|
1. Polyuric states
| 1. Decreased intake
|
2. Extracelluar fluid (ECF) volume expansion
| 2. Gastrointestinal losses
|
3. Acquired tubular dysfunction
| 3. Redistribution
|
4. Inherited renal Mg wasting disorders (Table 8–4) | 4. Lactation |
5. Medications Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|





