Fig. 24.1
Simplistic schematic representation of the general relationship between inflammatory cells (resident and infiltrating), vessels, fibroblasts, tubules and extracellular matrix in the normal kidney. The interrelation and interaction between these elements in response to a variety of insults may result in tubulointerstitial injury and chronic tubulointerstitial nephritis
Mechanisms of Tubulointerstitial Injury
The tubulointerstitium may be injured in a variety of ways. Examples include toxic insults (e.g. heavy metals), drugs (e.g. analgesics), crystals (e.g. calcium phosphate, uric acid), infections, obstruction, lipid deposition, immunologic mechanisms and acute elevations in capillary pressure and by ischemia [5–7]. Glomerular disease can also injure the tubulointerstitium through mechanisms that may involve glomerular cytokine release [8], direct effects of proteinuria on the tubules [9] or ischaemia [6], amongst others.
Experimental models of renal disease provide insights into possible processes responsible for these changes. Proteinuria has been found to be a major mechanism for initiating tubular injury; studies suggest that this may be due to cytokines and pro-oxidant molecules present in the proteinuric urine [9], as well as complement components that are then activated to form the complement membrane attack complex (C5b-9) which in turn can directly activate the tubular cells [10]. Ischaemia induced by intrarenal vasoconstriction and/or loss of peritubular capillaries also leads to tubular injury and fibrosis that is often most severe in the outer medulla and juxtamedullary regions where the kidney normally has borderline oxygen saturation [11].
Response to Trauma
While the type and size of injury are important, the kidney’s response is surprisingly universal, resembling the generic wound healing process that occurs in all organs [12, 13].
The tubulointerstitial reaction to trauma begins, as elsewhere, with inflammation. In the case of physical trauma, platelet aggregation forms a hemostatic plug, acts as a provisional matrix and initiates an inflammatory response. Likewise, in immunological and toxic injuries, the immune response triggers the recruitment of inflammatory cells. Injuries to the tubules and peritubular capillaries are often primary events and result in the release of chemotactic substances (chemokines and lipid factors) and expression of leukocyte adhesion molecules (such as ICAM-1 and osteopontin). Tubular cells also express HLA antigens and secrete complement components and vasoactive mediators, which may further stimulate or attract inflammatory cells. Tubular cell proliferation and hypertrophy are part of an early attempt to regenerate and repair injured tissue.
Antibodies have been used to histochemically identify the infiltrating haematogenous cells in various forms of inflammation. Important cells involved in the process include haematogenous cells and connective tissue cells such as resident macrophages, lymphocytes and fibroblasts. In the early stages of acute inflammation, polymorphonuclear granulocytes (polymorphs) are the predominant infiltrating leucocytes. If the tissue is infected by pus-producing bacteria, there is sustained and enhanced polymorph infiltration. The intensity of this response subsides as polymorphs are lost though apoptosis. In chronic interstitial nephritis, the interstitial infiltrate is similar in nearly all forms of tubulointerstitial injury [14]. T lymphocytes form the majority of haematogenous cells present, with monocytes, B cells, natural killer cells and plasma cells making up the remainder. Within the T-lymphocyte population, there is variability in the relative proportion of T-cell subsets [14].
Monocyte infiltration into extravascular tissue is a key event. Following activation, macrophages also secrete a wide range of biologically active mediators which are involved in tissue destruction (proteases, oxygen-derived free radicals), chemotaxis (cytokines, chemokines), vascular haemodynamics (thromboxane A2, prostaglandins) and production of matrix (growth factors, “remodelling” collagenases) [15]. There is, however, heterogeneity amongst macrophage populations, with different stimuli producing different activation states. Macrophage accumulation may persist with their numbers supplemented by local proliferation [16] and re-circulation [17].
What follows in chronic interstitial nephritis is an attempt at wound healing, a universal response to the inflammation that follows injury. It consists of a series of consecutive but overlapping events: these include cell proliferation, migration, extracellular matrix deposition (collectively known as fibrogenesis), resolution and remodelling.
Fibroblasts are the principal cells in fibrogenesis; selective deletion of fibroblasts by transfection with a so-called death gene is sufficient to prevent experimental renal interstitial fibrosis after injury [18]. The paucity of specific markers for fibroblasts, however, has meant that they have been difficult to identify. Activated fibroblasts (myofibroblasts), however, can be localised from their expression of alpha-smooth muscle actin, an actin isoform usually only found in vascular smooth muscle cells. The origin of (myo)fibroblasts in chronic tubulointerstitial nephritis is controversial. Rapid proliferation of resident fibroblasts seems to be an important early event in response to injury, mitogenesis exponentially increasing myofibroblasts at the site of renal injury within days [19]. Fibroblasts may also be derived from tubular epithelia in a process termed epithelial–mesenchymal transition [20], from circulating precursors [21], and after migration from adjacent perivascular areas. It remains to be seen if these processes are temporally distinct in fibrogenesis.
In each case, fibroblasts are the main source of the extracellular matrix proteins, mostly collagen and fibronectin, which provide structural integrity, while the myofibroblast provides force for wound contraction. Importantly, fibrogenesis continues as long as these cells persist in the wound, their removal or loss by apoptosis being part of the process that limits the extent of scarring.
The process of fibrogenesis is regulated by a plethora of cytokines and growth factors. Transforming growth factor-β (TGF-β) and platelet-derived growth factor (PDGF) have consistently been shown to be important in a variety of situations [5]. Conversely, a number of naturally occurring renoprotective factors act as a counterbalance and limit fibrogenesis. The most studied of these, hepatocyte growth factor (HGF) and bone morphogenic protein 7 (BMP7), downregulate pro-fibrotic TGF-β signalling by interfering with signal transduction [22].
Finally, the matrix is subject to remodelling by collagenous and non-collagenous proteinases, including metalloproteinases and plasmin/plasminogen family. Although the functional significance of these proteases is poorly understood, it is widely thought that the balance between collagen synthesis and degradation is an important factor in determining the extent of matrix accumulation [23].
The inflammatory and wound healing response is therefore an attempt to repair injury and restore tissue function. However, the tubulointerstitium has a very limited capacity to regenerate after a prolonged insult. Whereas acute wounds go through this linear series of events, chronic non-healing wounds do not. Some areas of chronic wounds are in different phases at the same time, and progression to the next phase does not occur in the same co-ordinated manner. Loss of vascular perfusion in this area may result in hypoxia, further exacerbating the failure of repair. The end result of chronic interstitial nephritis is the failure of wound healing and accumulation of excess matrix, so-called pathological scarring (Fig. 24.2).
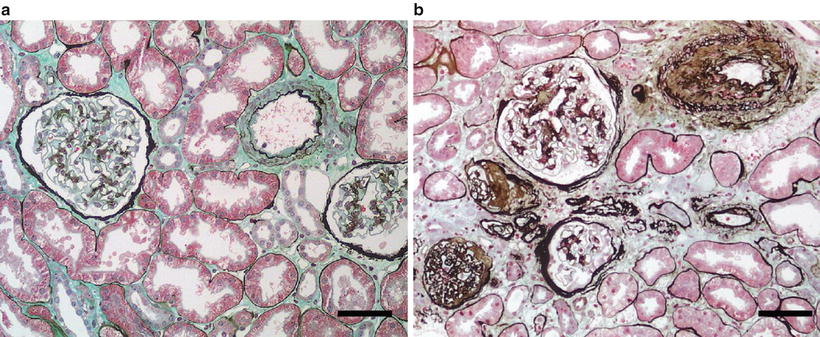
Fig. 24.2
(a) Normal kidney. (b) Tubulointerstitial disease with tubular atrophy and tubular basement membrane thickening, perivascular fibrosis, and glomerular sclerosis and interstitial fibrosis [Silver methenamine/Masson’s trichrome stain; scale bar = 100 μm (microns)]
Epidemiology
Although chronic TIN characterises progressive renal disease of all aetiologies, idiopathic forms of chronic TIN account for relatively few patients reaching end-stage renal disease (ESRD). For instance, of the primary renal disease diagnoses in the incident cases (n = 2,210) recorded in 2005 in the Australia and New Zealand Dialysis and Transplant Registry, only 130 (6 %) had an “uncertain diagnosis”. Of the remaining cases (n = 1,880), importantly only 57 cases (<3%) were recorded where the primary insult was attributed to the interstitium [24].
If one includes those particular conditions where the kidneys are macroscopically abnormal, such as reflux nephropathy (3 %), analgesic nephropathy (3 %), obstructive uropathy (2 %) and cystic renal disease (7 %), and where the major histologic lesions are also “tubulointerstitial”, this expands the proportion of TIN causes of ESRD towards 20 %. Reports from other areas of the world have also indicated quite a variable incidence of chronic tubulointerstitial disease in patients with ESRD, with a range from 42 % in Scotland to 3 % in the USA [25]. The marked variability in incidence may relate to differences in how diagnoses are made and in particular the use of renal biopsy, differences in aetiologies and toxin/drug exposure and different available measures to prevent or treat the various conditions.
Clinical Manifestations
Most patients with primary chronic TIN will exhibit proteinuria in the sub-nephrotic range. The urinary sediments will be relatively “inactive” in appearance. White blood cells and tubuloepithelial cells may be observed, and rarely white blood cell casts or tubuloepithelial cell casts may be present. If measured, a (partial) urinary concentrating defect is often apparent (perhaps with accompanying polyuria and nocturia), which may occasionally be severe enough to result in a nephrogenic diabetes insipidus. Other tubular defects occur, which may relate to the main site of injury, resulting in either proximal tubule defects (with aminoaciduria, glycosuria, phosphaturia, proximal renal tubular acidosis [RTA] or, rarely, Fanconi’s syndrome) or distal tubular defects (especially type IV RTA). Although many diseases affecting the tubulointerstitium are also associated with the inability to conserve salt on a low-salt diet (resulting in salt-wasting syndromes), there is evidence that certain chronic tubulointerstitial diseases, particularly if they are associated with microvascular disease, may also be associated with a relative inability to excrete salt, especially when the patient has a relatively high-salt diet [26]. This apparent phenomenon probably explains why some tubulointerstitial diseases, such as lead nephropathy, gouty nephropathy, cyclosporine nephropathy and analgesic nephropathy, are frequently associated with salt-sensitive hypertension. Thus, in summary, primary or idiopathic and acquired tubulointerstitial diseases may as a consequence present problems of either salt excretion or salt conservation [26].
Pathology
Since progressive interstitial disease (particularly interstitial fibrosis) is part of a common final pathway for all causes of renal disease [1], the identification of any specific histologic characteristics in chronic TIN is problematic. In effect, all of the major histologic features of TIN are “nonspecific”. The pathological diagnosis of primary TIN is often established rather by the absence of specific glomerular or vascular features, thus pointing to the tubulointerstitium as the likely area of primary insult in the pathologic process. Notably, as progressive tubulointerstitial injury occurs, the glomeruli may become secondarily involved, with hyalinosis and glomerular sclerosis occurring as a consequence of glomerular hypertension and so-called hyperfiltration injury.
Interstitial Fibrosis and Tubular Atrophy
Interstitial fibrosis is a fundamental histologic feature of chronic TIN and is characterised by an increased interstitial space relative to glomerular, vascular and tubular volume. This is due in large part to the accumulation of excessive connective tissue. The pattern may vary and be either focal or diffuse depending on the nature of the original insult.
Likewise, tubular atrophy is a nonspecific lesion. Tubular atrophy is observed frequently in areas of developing interstitial fibrosis. The tubular basement membrane (TBM) is often thickened; tubules may be dilated or atrophic and are often separated from each other by dense interstitial fibrosis. In some conditions (e.g. lithium-associated nephropathy), the degree of tubular atrophy can be correlated with a measurable renal functional abnormality such as impairment of urine-concentrating ability or impairment of urinary acidification.
Fibrosis on histology represents a disproportionate amount of ECM and may be due to either an increased deposition of ECM or the atrophy of renal parenchyma [27].
Glomerular Sclerosis
Glomerular sclerosis is accepted as occurring secondarily to the tubulointerstitial insult. The normal glomerular structure is replaced eventually by global fibrosis. In progressive renal disease with associated hyperfiltration, surviving glomeruli may be observed to be normal or show focal and segmental hyalinosis lesions, as might be the case in any renal disease under such conditions.
Occasionally glomeruli are structurally well preserved but are non-functional due to periglomerular fibrosis that effectively prevents outflow of the urine into the proximal tubules (“atubular glomeruli”) [28]. Disruption of the tubular segments may be the result of tubulointerstitial injury and lead to nephron loss which can also occur, or tubulointerstitial injury may indirectly lead to periglomerular fibrosis that constricts the outflow of the urine from Bowman’s space into the proximal tubule, again leading to the development of “atubular glomeruli”.
The inflammatory cells of the renal interstitial infiltrate (macrophages and lymphocytes) may also release oxidants and vasoactive compounds such as angiotensin II that can lead to local vasoconstriction and modulate glomerular hemodynamics and impair sodium excretion. Indeed, there is evidence that they may contribute to the pathogenesis of some forms of salt-sensitive hypertension [29].
Cast Formation
The presence of casts within tubular lumina is also a common and nonspecific finding. The casts are usually strongly PAS positive and are largely composed of Tamm–Horsfall protein but may also contain desquamated tubuloepithelial cells embedded in the Tamm–Horsfall protein as well as other products such as immunoglobulins [30]. Occasionally the casts take on a homogenous waxy appearance in dilated tubules, suggesting a chronic obstructive element to the tubulointerstitial injury.
Interstitial Cell Infiltrate
Interstitial cell infiltrate is not a consistent finding. Cells in the infiltrate include lymphocytes and monocyte–macrophages. Depending on the aetiology of the tubulointerstitial disease, there may be other cell types such as neutrophils, eosinophils or plasma cells. Cells infiltrating into the interstitium are usually observed in patches (focal) but may be more diffuse.
Loss of Peritubular Capillaries
Again it has long been recognised that a loss of peritubular capillaries parallels progression. It is important to remember, however, that this reduction in capillaries may be both causative and a consequence of increased ECM accumulation.
Aetiologies
Analgesic Nephropathy
Definition and Epidemiology
Analgesic nephropathy was first recognised as a clinical entity as early as the 1950s in Swiss watch factory workers, who were habitually taking large amounts of over-the-counter analgesics, particularly combination analgesics containing phenacetin. Shortly thereafter analgesic nephropathy was recognised as an important cause of ESRD, especially in certain areas of Europe and Australia. In two Australian States, Queensland and New South Wales, as many as 25 % of patients with ESRD in the 1970s had analgesic nephropathy identified as the primary disease, and in the 1980s in Belgium the prevalence was nearly 20 % [31]. In contrast, analgesic nephropathy was substantially less frequently diagnosed in the USA, which presumably related to a lesser clinical awareness of the entity as well as the lower availability of compound analgesic mixtures, particularly those containing phenacetin.
The eventual recognition of compound analgesic mixtures particularly those containing phenacetin as a risk factor for renal disease importantly led to the prohibition of such compounds in a number of countries. Whereas the restriction of compound analgesic sales in Scandinavian countries Finland, Canada, and the UK led to variable effects on the incidence of ESRD, in Australia a very significant decline in the incidence of ESRD related to analgesic nephropathy was observed [32]. Following the introduction of the legislation, the number of new patients commencing dialysis fell significantly from 1980 (Fig. 24.3). Controversy has continued as to whether the reduction in incidence related to the banning of the compounds rather than the elimination of phenacetin from the mixtures [33].
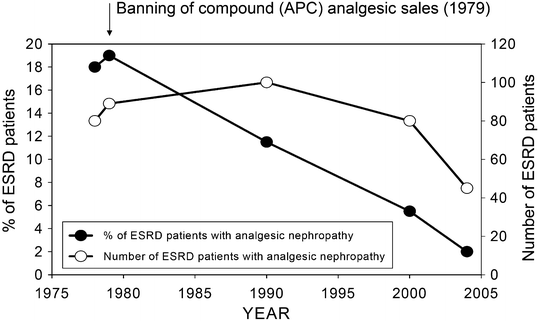
Fig. 24.3
Relationship between the incidences of ESRD attributed to analgesic nephropathy in Australia between 1978 and 2005. Graph compares the total number of patients with analgesic nephropathy as the diagnosis and the proportion of patients with ESRD for whom the diagnosis was analgesic nephropathy. Note that the sale of aspirin–phenacetin–codeine (APC) compounds was banned in 1979. Data from Australia and New Zealand Transplant Registry (ANZDATA) registry reports for 1979–2005, ANZDATA, Adelaide, Australia
The decline in patients requiring dialysis was most pronounced for those between the ages of 40 and 49 years. In the early 1990s, the percentage of hemodialysis patients with analgesic nephropathy as an incident disease was 9 % in Australia, 3 % in Europe and 0.8 % in the USA [31]. Analgesic nephropathy as a presenting cause of ESRD in Australia accounts currently for only 2.8 % of cases [24] (Fig. 24.3).
So in summary, the withdrawal of phenacetin was not associated with a complete eradication of analgesic nephropathy, but it was associated with a marked diminution in the incidence of ESRD attributed to analgesic nephropathy. Considerable evidence (experimental, pharmacologic and epidemiologic) [34, 35] evolved, suggesting that succeeding analgesic mixtures not containing phenacetin but containing compounds such as acetaminophen, pyrazolones and even aspirin [36] were capable of producing the typical kidney pathology lesions (see below).
Aetiologic Agents
The clinical syndrome of classical analgesic nephropathy resulted from the prolonged misuse or abuse of compound analgesics containing aspirin or antipyrine, combined with phenacetin, acetaminophen (paracetamol) or salicylamide, and caffeine or codeine. Lately, it has been suggested that a similar lesion can be induced with chronic nonsteroidal anti-inflammatory agent (NSAID) use. NSAIDs reported to induce analgesic nephropathy include alclofenac, antipyrine, benoxaprofen, fenoprofen, ibuprofen, indometacin (indomethacin), mefenamic acid and naproxen; there are also reports suggesting aspirin and acetaminophen may have contributory roles. While the role of non-phenacetin compounds in causing analgesic nephropathy remains controversial [31], there is some evidence that the chronic use of aspirin or acetaminophen may accelerate renal progression of any aetiology [37]. In this regard, it is important to recognise that agents that block intrarenal prostaglandin synthesis, such as NSAIDs, can be acutely nephrotoxic and can cause an acute but not always reversible decline in glomerular filtration rate (GFR) or acute tubular necrosis, especially in the setting of volume depletion, in the elderly and in diabetics [38–41]. Acute papillary necrosis may also occur particularly if large doses are ingested, and acute renal papillary necrosis induced by NSAIDS has certainly been demonstrated in animal studies [42]. NSAIDs may also be idiosyncratically associated with a minimal change-like lesion glomerular with acute presumably allergic TIN.
Establishing a link between NSAIDS and chronic kidney lesions is problematic. The data is scarce and epidemiological in nature. The studies have been criticised for poor study design, and the odds ratio for risk of end-stage renal disease varies from decreased risk (RR = 0.6) to markedly increased (RR = 10) depending on duration of exposure to NSAIDS [43, 44], age and gender and adjustment for other analgesics.
Because of the uncertainties surrounding toxicologic data and pathogenesis, it follows that it is difficult to establish the critical amounts and periods of intakes of the various analgesic agents required to produce analgesic nephropathy. Epidemiologic studies generally show a dose-dependent risk for developing analgesic nephropathy with compound analgesics, especially with those containing phenacetin, and it has been estimated that there is a 15- to 20-fold increased relative risk for the development of chronic renal disease when the total consumption of phenacetin exceeds 1,000 g [45]. For NSAIDS, an association was demonstrated between prolonged exposure (up to 26,000 doses) and the demonstration of renal papillary necrosis on imaging [46] but with only modest effects on kidney function.
Limitations of study design and of diagnosis have made the interpretation of case–control and other epidemiological studies [43, 47–49] highly problematic [50]. At the current time, it has not been possible either to establish a clear link between prolonged analgesic exposure and progression of underlying chronic kidney disease of any aetiology or to reach a clear understanding of the risk of end-stage kidney disease associated with analgesic use. Interestingly, despite the widespread perception that analgesic use may promote progressive CKD, a recent analysis from the Danish National Registry of over 6,600 ESRD patients reported that over one-third received NSAIDS for a median of 40 days in the 3 years before starting dialysis [51].
Pathogenesis and Pathology
The principal site of renal injury with chronic analgesic abuse is in the medulla and papillae, sites that are vulnerable because of the concentration of toxic metabolites built up by the countercurrent mechanism and because of the low oxygen tension present in these anatomical regions. The injury may relate to the net effects of several metabolites [52]. Phenacetin, for example, is converted to acetaminophen, which can deplete cells of glutathione and result in the generation of oxidative and alkylating metabolites. Aspirin [53, 54] and NSAIDs [55, 56] can result in the reduction of vasodilatory prostaglandins; caffeine may be metabolised to adenosine, with vasoconstrictive effects within the kidney. Collectively, these substances may lead to oxidant and ischemic injury to the medulla and papillae, an effect that is exacerbated in the setting of volume depletion [57]. Renal papillary necrosis with features of an ischemic infarct may develop, with atrophy of the overlying cortex leading to chronic interstitial changes.
The quintessential pathologic lesion of analgesic nephropathy is renal papillary necrosis [58], a coagulative necrosis involving the medulla, including the loops of Henle, the vasa recta, the medullary interstitial cells and the collecting ducts. The cortical changes of chronic interstitial nephritis overlying the necrotic papilla are secondary and comprise tubular atrophy, interstitial fibrosis and a mononuclear cellular infiltrate. The presence of a golden-brown lipofuscin-like pigment in tubular cells, and the characteristic analgesic microangiopathy or capillary sclerosis, is highly indicative of an analgesic aetiology. Glomerular changes of focal glomerular sclerosis and hyalinosis associated with proteinuria are similar to those described in reflux-associated nephropathy and tend to be late features.
Clinical Features
The vast majority of patients are female [59] and have a history of chronic pain syndromes and other somatic complaints or symptoms suggestive of a broader analgesic syndrome [60]. An addictive and dependent personality trait characterised by introversion, psychoneurosis and an external locus of control has been reported, and an association with cigarette smoking has been noted. Peptic ulcer disease and gastrointestinal symptoms are also present in many patients. Denial of analgesic abuse is common.
The renal function abnormalities of analgesic nephropathy (other than a slowly progressive impairment of GFR) include impaired urine-concentrating ability (often an early defect possibly associated with polyuria), urinary acidification defects and impaired sodium conservation [54]. The urinalysis frequently shows pyuria with or without urinary tract infection, micro- or macrohaematuria may be present, and non-nephrotic range proteinuria. Proteinuria occurs in at least half of the patients [61] and increases progressively as GFR decreases with advanced disease. The proteinuria is of both tubular and glomerular origin, the latter as a consequence of secondary glomerular sclerosis that develops as renal function deteriorates. Hypertension is a very common clinical feature [54].
Although patients with any form of chronic kidney disease are at greatly increased risk of cardiovascular mortality, cardiovascular disease, fatal or nonfatal myocardial infarction, heart failure or stroke, patients with analgesic nephropathy may be at even greater risk for premature atherosclerosis [62]. Atheromatous renal artery stenosis is also more common in analgesic abusers. The increased atherogenic tendency probably relates to multiple factors, including hypertension, smoking and hyperlipidemia, and perhaps the formation of atherogenic oxidised low-density lipoproteins under the oxidative influence of phenacetin [63].
An important association of analgesic nephropathy is the increased risk of transitional cell carcinoma of the uroepithelium (renal pelvis, ureter, bladder and proximal urethra) [64]. This should be especially considered in patients with gross haematuria.
Diagnosis
The disease is suggested by a history of analgesic abuse coupled with urographic, sonographic and/or tomographic findings showing papillary calcifications and bilateral atrophic but often asymmetric kidneys with irregular contours (“bumpy”). The imaging findings are not always easily distinguishable from those of reflux-associated nephropathy. Papillary calcification is a very helpful diagnostic feature and may be best detected by a noncontrast CT scan [65]. The differential diagnosis of papillary necrosis, the key underlying pathology in analgesic nephropathy, includes diabetic nephropathy, sickle-cell nephropathy and, very rarely, obstructive uropathy and reflux-associated nephropathy. Papillary deposits of calcium can also be seen in medullary sponge kidney and all forms of nephrocalcinosis.
Treatment
Management consists of avoiding phenacetin-containing analgesic mixtures (which are largely not available now) and reducing or ideally completely stopping ingestion of other analgesic medications [66, 67] as no specific treatments are available. Management is essentially similar to that for all patients with chronic kidney disease, including careful monitoring of blood pressure and the use of renoprotective agents such as ACE inhibitors for those with impaired kidney function (stage 3 CKD with an eGFR <60 mL/min/1.73 m2). Because of the increased incidence of uroepithelial tumours, long-term follow-up is necessary. A cause of flank pain may be obstruction due to detached (necrotic) papillae lodging in the ureter. In the presence of infection, this complication can be a life-threatening clinical scenario.
Lithium Nephropathy
In the mid-1970s, it was recognised that long-term administration of lithium salts to patients with severe unipolar and bipolar affective illness may be associated with the development of chronic kidney disease. Several different forms of renal injury were identified [68]. It should be emphasised, however, that because the therapeutic index of the lithium ion is narrow, the most important complication of short- or long-term lithium administration remains the development of acute lithium intoxication. The kidneys are effectively exclusively responsible for the excretion of lithium and, therefore, are pivotal to the development of this important complication.
Lithium-Associated Polyuria, Polydipsia and Nephrogenic Diabetes Insipidus
Lithium ingestion is associated with the development of resistance to vasopressin (antidiuretic hormone), resulting in polyuria (usually defined as a urine volume exceeding 3,000 mL/24 h) and polydipsia in up to 40 % of patients. Lithium is the most common cause of iatrogenic diabetes insipidus [69–74]. Lithium accumulates in the collecting tubule cells after entering these cells through sodium channels in the luminal membrane. It then interferes with the ability of vasopressin to increase water reabsorption [75] by inhibiting adenylate cyclase and hence cyclic AMP (cAMP) production and also by decreasing the apical membrane expression of aquaporin 2 [76], the collecting tubule water channel. Although this common side effect is widely regarded as reversible, lithium-induced impairment of urine-concentrating ability was shown in some patients to persist many months after cessation of lithium therapy. Interestingly, lithium is also less commonly a cause of hypercalcemia. This complication is a consequence of an increased release of parathyroid hormone (PTH) and is associated with local (parathyroid) cAMP production. Persistent hypercalcemia could potentially exaggerate the tubular concentrating defect and contribute to the development of chronic interstitial nephritis in lithium-treated patients (see section “Hypercalcemic Nephropathy”).
A lithium-induced impairment in distal urinary acidification [type 1 (distal) RTA] may be seen in parallel with nephrogenic diabetes insipidus with lithium therapy. A lithium-induced impairment in distal urinary acidification [type 1 (distal) RTA] is a partial functional defect that is rarely of clinical importance. A lithium-induced decrease in the activity of the H+ ATPase pump in the distal nephron (collecting ducts) may be the main cause of this defect.
Acute Lithium Toxicity
Acute impairment of GFR and tubular injury (acute tubular necrosis) associated with episodes of acute lithium intoxication have for many years been recognised potential complications of lithium therapy. The risk is greatly increased in the presence of volume depletion [77].
Histologically, the most distinctive change associated with lithium occurs in the distal convoluted tubules where there is ballooning, swelling and vacuolation of the cell cytoplasm, accompanied by strands and granules of periodic acid-Schiff (PAS)-positive staining material (glycogen). This lesion is not only acute but also quite reversible [78, 79].
Chronic Lithium Nephropathy
A new observation in the 1970s was the recognition that, in lithium-treated patients, the polyuria (and polydipsia) was not always reversible [80–86].
A large number of studies subsequently demonstrated a correlation between the duration of lithium therapy and persistent impairment of urine-concentrating ability [68] (Fig. 24.4). Biopsies in these patients subsequently revealed primarily a focal chronic interstitial nephropathy with interstitial fibrosis and tubular atrophy, and more recently the degree of associated glomerular sclerosis has been recognised [87].
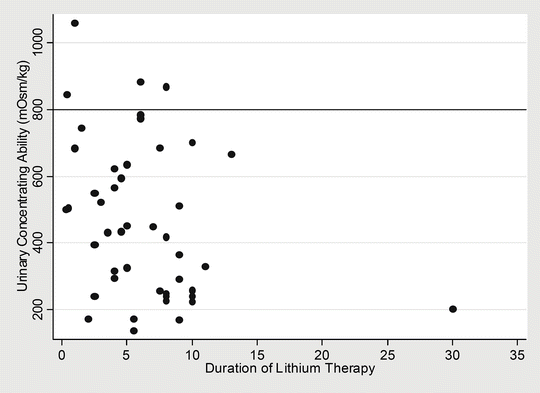
Fig. 24.4
Relationship between urinary concentrating ability (measured after overnight fluid restriction in mOsm/kg) to duration of lithium therapy in 46 of 50 patients on long-term maintenance lithium for unipolar and bipolar affective disorders. Note that only very few patients with short durations of lithium therapy achieved urinary concentrations above 800 mOsm/kg. Correlation coefficient; r = −0.5200 (p-value < 0.001). Adapted from Walker [68], with permission
Another characteristic finding is the presence of microcystic changes in the distal tubule. More recently, these cystic changes have been well demonstrated using MR imaging [88]. Patients at particular risk seemed to be those with severely impaired urine-concentrating defects and those with a history of repeated episodes of acute lithium intoxication.
Interestingly, most patients with chronic lithium nephropathy have a relatively well-preserved GFR [73, 89–92] in relation to the distal tubular defect(s). Although a small number of patients have chronic kidney disease with renal impairment, in most studies there is no correlation between GFR and the duration of lithium therapy (unlike with the concentrating defect). At least in one study, the slow rate of deterioration of renal function and final creatinine clearance was related to duration of therapy [93]. In patients taking lithium who do not have repeated episodes of acute lithium intoxication, the role of lithium in chronic interstitial disease remains controversial. Nonetheless, the number of reported cases of either serious renal insufficiency or chronic kidney disease continues to cause concern as there are clearly cases of elevated serum creatinine (>177 μmol/L or >2.0 mg/dL) with no obvious cause other than the long-term ingestion of lithium. In Lepkifker et al.’s review [94], it was estimated that 20 % of patients on long-term lithium develop renal insufficiency [70, 94–96]. Controversy also persists because although differences in urinary concentrating ability between patients on lithium and patients not on lithium are apparent, patients with affective disorders do not concentrate urine normally [97] (Fig. 24.5). Furthermore, other similar degrees of functional change [97, 98] and chronic interstitial change, including interstitial fibrosis, have been noted in patients with affective disorders not being treated with lithium [99], although the characteristic microcystic dilatation of the distal tubules is generally absent. The role of other psychotropic medication in this setting is unknown. If chronic TIN does occur in association with long-term stable maintenance therapy with lithium, it probably requires many years of therapy [93, 100, 101] and the consequent degree of renal insufficiency, as indicated above, is likely to be relatively mild. Perhaps the most important factor in preservation of renal function and histology is prevention of episodes of acute intoxication.
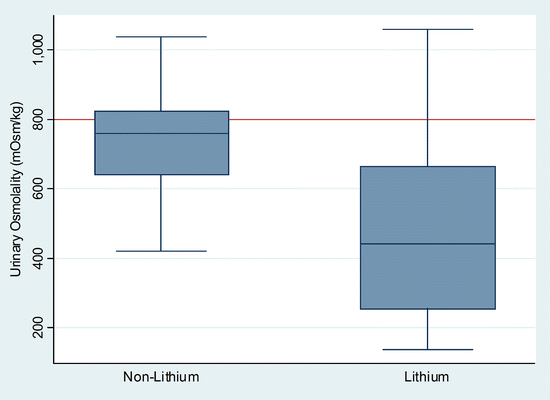
Fig. 24.5
Box plot quartiles of urinary concentrating ability (measured after overnight fluid restriction in mOsm/kg) in 46 (of 50) patients (Lithium) on long-term maintenance lithium for unipolar and bipolar affective disorders compared to 27 (of 32) patients with affective disorders not on lithium (non-lithium). Note the number of non-lithium patients with urinary concentrations below 800 mOsm/kg (adapted from Walker RG, Doctoral Thesis “Lithium Nephrotoxicity”, 1986, University of Melbourne)
Management of Lithium-Associated Renal Disease
After eliminating other possible causes of polyuria and polydipsia [104], especially psychogenic polydipsia (or potomania), one should first consider reducing the dose of lithium, aiming for therapeutic concentrations (12-h serum lithium concentrations) of 0.4–0.6 mmol/L (0.28–0.42 mg/dL) or converting to a single daily dose [105] aiming for a lower trough serum lithium concentrations. For prophylaxis, levels of 0.4–0.6 mmol/L (12 h standard trough serum lithium concentrations) may be sub-therapeutic in a proportion of patients [106], compared to recommended levels of at least 0.5–0.8 mmol/L (identical in patients with bipolar and unipolar illness) [107], but also likely to be associated with fewer side effects. Values of 0.8–1.0 mmol/L are likely to have marked efficacy but many side effects. The potassium-sparing diuretic amiloride can also reduce the urine output by up to 50 % and has the added advantage of blocking lithium uptake through the sodium channels in the collecting duct [108]. Amiloride is most effective when the concentrating defect is mild and potentially reversible and has generally been disappointing in patients with severe defects in urine-concentrating ability. Although thiazide diuretics are effective in treating lithium-induced polyuria, they should be avoided as they pose risks for the induction of acute lithium intoxication because the resultant volume contraction causes an increase in sodium and lithium reabsorption in the proximal tubule. Avoidance of NSAIDs if possible is also recommended.
A practical approach to the patient on long-term lithium treatment should include a regular (at least yearly) estimation of renal function measured as serum creatinine (or estimated GFR), estimation of spot urine albumin/creatinine ratio and measurement of 24-h urine volume [95, 107, 109, 110]. Because progressive renal injury with a reduced GFR (or raised serum creatinine) in a patient without prior acute lithium intoxication is relatively unusual, a raised serum creatinine should probably in the first instance be treated with dose reduction. For persistently elevated serum creatinine estimations, a renal biopsy could be considered, although the result would not often lead to a recommendation to cease lithium entirely. At all times, the risk of discontinuation of lithium in a patient with a severe unipolar or bipolar affective disorder needs to be measured against the minimal risk of progressive renal injury. As for analgesic nephropathy, the management of lithium-associated nephropathy with chronic kidney disease is similar to that for all patients with chronic kidney disease and includes careful monitoring of blood pressure and the use of renoprotective agents such as ACE inhibitors for those with impaired kidney function (stage 3 CKD with an eGFR <60 mL/min/1.73 m2). Care is required when introducing any new agent to a patient taking lithium to check serum lithium estimations because of the potential of some agents to acutely alter GFR and/or renal clearance of lithium.
Also, a range of other therapies exist for bipolar and unipolar affective disorders including anticonvulsants such as carbamazepine, valproate and lamotrigine, which may be alternative medications in cases of severe lithium-induced renal symptoms or renal insufficiency.
Heavy Metals
Lead Nephropathy
Acute lead intoxication is rare but may present with abdominal pain, encephalopathy, haemolytic anaemia, peripheral neuropathy [111, 112] and proximal tubular dysfunction (as manifested by either a proximal RTA or Fanconi’s syndrome) [113]. In contrast, chronic lead intoxication, which may develop insidiously and even follow decades after acute lead poisoning [114], is much more subtle with impaired renal function, episodic gout (“saturnine”) and hypertension being the main manifestations [115]. Occasionally, patients may exhibit other signs of chronic lead intoxication including peripheral motor neuropathies, anaemia with “basophilic” stippling and perivascular cerebellar calcifications [116]. Individuals known to be at risk from lead intoxication include children exposed to lead-based paints, adults drinking “moonshine” and residents living close to lead-smelting factories and other industries expelling lead into the environment [117, 118].
Tubular functional disturbances are usually the earliest manifestations of chronic lead nephropathy leading to hyperuricaemia (resulting from diminished urate secretion), aminoaciduria or renal glycosuria. Chronic exposure can ultimately lead to a chronic interstitial nephritis. The pathogenesis of the renal disease may be related to the proximal tubule reabsorption of filtered lead, with subsequent accumulation in the proximal tubular cells. Histologically, proximal tubular injury, with intranuclear inclusion bodies composed of a lead–protein complex, may be observed initially but with prolonged lead exposure the major histologic features of chronic interstitial nephropathy: progressive tubular atrophy and interstitial fibrosis associated with chronic renal insufficiency occur in the presence of a relatively normal urinalysis, but associated with hypertension, and gout [119]. Populations with high rates of diabetes, hypertension and/or chronic kidney disease appear to be at greater risk of developing adverse renal effects from lead [118]. Ideally, all patients with significant hyperuricaemia and renal insufficiency should have a history of occupational lead exposure excluded [120], as confusion may occur with chronic urate nephropathy in which urate deposits (tophi) may form in the renal interstitium.
Several epidemiologic studies in the general population have also suggested that low-level lead exposure may be associated with chronic kidney disease with renal impairment and/or hypertension. In these studies, an increase in lead burden has been shown, but no causal association has been established [121–123], leading to some investigators to question whether chronic lead exposure truly promotes renal failure in recent years [124].
Treatment
In the industrial and occupational settings including foundry workers and individuals working with lead-based paints and glazes, preventative measures to minimise exposure and low-level absorption are essential. In established cases, removal of the source of lead is obviously important. Otherwise, treatment involves the use of infusions of CaNa2 EDTA. The likelihood of a satisfactory response to CaNa2 EDTA chelation will be influenced by the degree of already established interstitial fibrosis.
Cadmium
Cadmium is a metal used in a variety of industries, especially in the manufacturing of alloys and electrical equipment. Historically, a major outbreak of cadmium toxicity including nephrotoxicity occurred in Japan as a result of industrial contamination of the Jinzu River in the Toyama prefecture, leading to contamination of rice crops. The disease, named itai-itai [127, 128] or “ouch-ouch”, primarily affected older women and was characterised by proximal tubular dysfunction [129], anaemia, severe osteomalacia and, rarely, progressive chronic interstitial disease [130]. Functional changes accompanying cadmium nephropathy include proteinuria, enzymuria, aminoaciduria, glycosuria, polyuria, hypercalciuria and increased urinary uric acid and of course urinary cadmium [131]. Cadmium workers also have a higher incidence of hypertension and renal calculi (the latter caused by hypercalciuria). Cadmium accumulates in renal tubular lining cells [132] bound to a small (30 % cystine) protein metallothionein, which actually protects against nephrotoxicity by binding cadmium in a nontoxic form. Chronic interstitial fibrosis and renal functional abnormalities have been reported to occur when cadmium levels in the renal cortex exceed a concentration of ~200 μg/g [133].
Arsenic
Arsenic, present in insecticides, weed killers, wallpaper and paints, rarely causes renal disease. Chronic arsenic toxicity manifests most commonly as sensory and motor neuropathies, distal extremity hyperkeratosis, palmar desquamation, diarrhoea and nausea, Aldrich–Mees lines (linear white bands on the nails) and anaemia. However, both proximal tubular dysfunction [renal tubular acidosis (RTA)] and eventually chronic interstitial fibrosis may occur [136]. The diagnosis is established by demonstrating elevated urinary arsenic levels.
Radiation Nephritis
In “acute radiation nephritis”, oedema, hypertension (including occasionally accelerated hypertension), dyspnea, headache and nocturia are possible manifestations. Proteinuria may be marked, whereas the urine sediment is relatively unchanged. Anaemia (normochromic, normocytic or microangiopathic) is likely to be present as is mild-to-moderate impairment of kidney function. Progression to a “chronic” form of radiation nephritis may be a consequence of acute radiation injury, or “chronic radiation nephritis” may present as a more indolent process, with proteinuria and progressive chronic kidney disease and even ESRD. The incidence of new cases appears to be fortunately low. Considering all cases in the Australian and New Zealand Dialysis and Transplant Registry (ANZDATA Registry) in the last 10 years, there have been less than two per annum [24].
The precise pathogenetic mechanism of radiation nephritis remains obscure. Histologic features are similar between human and laboratory animals [137], and degenerative, inflammatory and thrombotic changes that appear early advance ultimately to severe glomerular sclerosis, tubular atrophy with associated thickening of the TBM and interstitial fibrosis. Interstitial disease may occur in the absence of glomerular lesions, which vary from milder mesangial cell changes (increased cells, increased matrix and mesangiolysis) to frank glomerular capillary and arteriolar necrosis [138], which may include evidence of thrombosis [139]. Capillary walls are thickened and may typically show “splitting”. Vessels may show patchy intimal proliferation and thickening, intimal fibrosis and fibrinoid necrosis [140]. Severe disease is characterised by progressive interstitial fibrosis [141] and the presence of interstitial inflammatory cells.
The similarities in certain pathologic characteristics of radiation nephritis with those of haemolytic uremic syndrome and thrombotic thrombocytopenic purpura (HUS/TTP), and the prevalence of the lesions of HUS and associated deterioration in renal function in bone marrow transplant recipients treated with combined radiotherapy and chemotherapy lead to speculation that endothelial cell injury, possibly leading to local intravascular coagulation, is one possible pathogenetic mechanism. Radiation nephritis has also been seen in bone marrow transplant recipients following total body irradiation and particularly in patients receiving cyclosporine [142]. Whether cyclosporine A enhances the development of radiation nephropathy is not established.
Prevention is the major therapeutic approach to radiation nephritis. The risk of developing this condition may possibly be minimised by proper shielding of the kidneys and/or by fractionating the total body irradiation into several small doses over several days. Minimising the total dose [143] of irradiation to the kidney to less than 20–30 Gy (1 Gy = 100 rad) is recommended, but no specific treatment is available for established radiation nephritis.
Balkan Nephropathy
Aetiology and Pathogenesis
Balkan (endemic) nephropathy is a slowly progressive form of chronic TIN found almost exclusively in some well-defined areas in Croatia, Serbia, Bosnia, Bulgaria and Romania [144]. Balkan nephropathy is further geographically localised to a few areas along the Danube’s tributaries in topographical regions characterised by plains and low hills that have generally a high humidity and high rainfall. Although described originally, more than 50 years ago [145], the precise aetiology of Balkan nephropathy has never been established. A variety of factors have been either considered or implicated, including genetic factors, environmental agents (such as trace elements [146] and fungal and plant toxins) and immune disturbances.
It has been argued that the inheritance of Balkan nephropathy, the familial nature of which has also been recognised for many years, is more likely to be polygenic and that the environment might markedly influence the manifestations of the genotype. In the endemic areas, likely affected individuals are villagers, but several members of one family (or household) across one or more generations may be affected with many unaffected households being present in the same area. An early Bulgarian genetic study favoured an autosomal dominant form of inheritance [147]. Later chromosomal breakage and spontaneous aberration studies demonstrated linkage to the 3q25 band on chromosome 3 [148–150], in both affected individuals and in healthy relatives of individuals with Balkan nephropathy considered to be at high risk.
Related posts:
 General Approach to the Diagnosis and Management of Glomerular Diseases
Childhood Onset Nephrotic Syndrome
General Approach to the Diagnosis and Management of Glomerular Diseases
Childhood Onset Nephrotic Syndrome
 Membranoproliferative Glomerulonephritis
Membranoproliferative Glomerulonephritis
 Thrombotic Microangiopathies: Thrombus Formation Due to Common or Related Mechanisms?
Thrombotic Microangiopathies: Thrombus Formation Due to Common or Related Mechanisms?
 Glomerular Disease in Pregnancy
Glomerular Disease in Pregnancy
 Anti-glomerular Basement Membrane Disease
Anti-glomerular Basement Membrane Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


