Chapter 40 Biliary atresia
Overview
Biliary atresia (BA) remains a somewhat elusive disease, with our understanding confined as it is to its infancy but yet with the origin of BA essentially unknown. BA is potentially fatal, certainly if left to run its natural course (Hartley et al, 2009), but it can be treated effectively with expeditious surgery in a high proportion of patients. For the remainder, liver transplantation is an option, and indeed BA remains the single most common indication for liver transplant in the pediatric population.
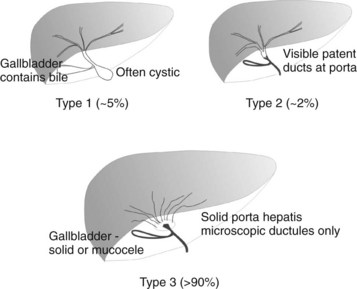
BA is an occlusive panductular cholangiopathy affecting both intrahepatic and extrahepatic bile ducts. The most common classification divides BA into three types based on the most proximal level of occlusion of the extrahepatic biliary tree (Fig. 40.1). In types 1 and 2, in which there is a degree of preservation of structure of the intrahepatic bile ducts, blunting, irregularity, and pruning occurs, and dilation may be absent, even when the duct is obstructed. In the most common type of BA, type 3, the intrahepatic bile ducts are grossly abnormal with myriad small ductules coalescing at the porta hepatis. When retrograde cholangiography is possible, this is seen as a “cloud.” In approximately 5% of cases, extrahepatic formation of cysts containing clear mucus or bile can occur in the otherwise occluded biliary tree. Such cystic biliary atresia (Caponcelli et al, 2008) is distinguishable clinically and histologically from simple obstruction in a cystic choledochal malformation, where there is preservation of an epithelial lining and retention of a normal and often dilated intrahepatic biliary tree.
Etiologic Heterogeneity
BA is not one disease, certainly not one with a single cause. In all probability it is a phenotype resulting from a number of different etiologies. Three groups can be defined clinically: 1) biliary atresia splenic malformation (BASM) syndrome (Davenport et al, 1993, 2008), 2) cystic BA (Caponcelli et al, 2008), and 3) isolated BA (Table 40.1).
Table 40.1 Etiologic Heterogeneity in Biliary Atresia
Other related entities are fewer in number, but there seems to be a relationship with other gastrointestinal anomalies, such as esophageal atresia and jejunal atresia, in a small proportion (<1% of large series); occasional cases show a defined chromosomal abnormality, such as “cat-eye” syndrome and chromosome 22 aneuploidy (Allotey et al, 2008).
Developmental biliary atresia is a term we use that includes BASM and cystic BA, for which the onset is almost certainly prenatal and is evident by the time of birth (Livesey et al, 2009). The onset of occlusion in isolated BA is much more contentious, and some authorities hold that the bile duct can be normal and patent at the time of birth and can then become occluded secondarily by virally mediated damage.
Biliary Atresia Splenic Malformation Syndrome
Although the association of BA with polysplenia had been recognized for some time, clarification of what constitutes BASM (Table 40.2) is only relatively recent (Davenport et al, 1993). The constellation of other anomalies is peculiar, and the reasons for this are still obscure. The common embryologic insult may simply be timing (30 to 35 days) rather than a specific genetic defect. Key genes are important in both bile duct development (JAG1, HNF-6 [ONECUT1]; Kohsaka et al, 2002) and visceral and somatic symmetry (INVS, CFC1; Bamford et al, 2000; Shimadera et al, 2007), although correlation with the human condition is patchy. A possible genetic link has recently been reported by a French group, who found an increased frequency of mutations in the CFC1 gene (on chromosome 2) compared with controls (Davit-Spraul et al, 2008).
Table 40.2 Spectrum of Anomalies in Biliary Atresia Splenic Malformation Syndrome
| Organ System | Malformation | Notes |
|---|---|---|
| Splenic | Polysplenia, double spleen (95%); asplenia (5%) | No evidence of immune hypofunction |
| Situs determination | Inversus (50%) | |
| Venous | Preduodenal portal vein (40%), portosystemic shunt (<2%); vena cava absence (50%) | |
| Intestinal development | Malrotation (60%) | |
| Cardiac | Atrial septal defect, ventricular septal defect, tetralogy of Fallot, etc. (~40%) | ± Dextrocardia |
| Liver | Mirror image, normal, or symmetrical | Position dictated by abdominal situs |
| Biliary appearance | Atrophic or absent gallbladder | Often noninflammatory remnants with absence of common bile duct and obvious segmental appearance |
| Pancreas | Annular (<5%) | |
| Miscellaneous | Immotile cilia syndrome, sacral agenesis |
Epidemiology
A clear difference in incidence is seen across the globe. The highest is reported in Taiwan, at one in 5000 live births (Hsiao et al, 2008). In Japan, the incidence is one in 10,000 (Nio et al, 2003), with national studies from the United Kingdom and France reporting an incidence of one in 17,000 and 19,000, respectively (Chardot et al, 1999; Livesey et al, 2009; Serinet et al, 2006). Although there are no national studies in North America, smaller regional studies suggest an incidence closer to that of Europe (Yoon et al, 1997) but with some interracial variation (The et al, 2007). Significant regional differences within the United Kingdom have also been observed, again ascribed to variations in racial origin and the nature of the BA. For example, developmental BA is more common in white infants than in those of Asian origin (Livesey et al, 2009).
A distinct female predominance in seen in those with developmental BA that is not seen in the isolated BA group for unknown reasons (Davenport et al, 1993, 2006; Livesey et al, 2009). A seasonal variation in incidence has also been observed in some regionally based studies (Yoon et al, 1997; The et al, 2007), although this has not been confirmed in the larger, national studies (Chardot et al, 1999; Livesey et al, 2009).
Pathophysiology
The nature of the mononuclear infiltrate has assumed a degree of importance, and many authors suggest this is the specific primary destructive element of BA (Davenport et al, 2001; Mack et al, 2007). Although not necessarily uniformly applied across the spectrum of BA, the infiltrate is largely composed of CD4+ T lymphocytes, specifically Th1 (Mack et al, 2004) and CD56+ natural killer (NK) cells (Davenport et al, 2001; Shivakumar et al, 2009), which exhibit markers for proliferation (CD71+) and activation (particularly LFA-1+ but also CD25+). Infiltrating CD8+ cells appear to lack markers of activation—such as perforin, granzyme B, and Fas ligand (Ahmed et al, 2001)—in contrast to other diseases such as primary biliary cirrhosis (see Chapter 9).
Expression of cell adhesion molecules, proteins involved in cell-cell binding, is an important precursor to an immune-active cell infiltrate. Both interstitial cell adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 are important in binding circulating leukocytes by interaction with cell-surface integrins, and they are expressed on sinusoidal and biliary epithelium of infants with BA (Dillon et al, 1997; Davenport et al, 2001).
Expression of soluble(s) ICAM-1 and sVCAM-1 in the circulation is also increased (Minnick et al, 1998; Davenport et al, 2005), as is seen in other immunologically mediated liver diseases such as primary biliary cirrhosis and sclerosing cholangitis; levels of sVCAM-1 at the time of presentation have been shown to be prognostic (Davenport et al, 2005). Increasing levels of various other cytokines (interleukin [IL]-2, tumor necrosis factor [TNF]-α, IL-18) can also be shown postoperatively (Fig. 40.2), but most discriminate poorly between patients who would clear their jaundice and those who would not; conversely, some cytokines (IL-2, interferon [IFN]-γ, IL-4, IL-10, TNF-α, and sICAM-1) were better predictors of subsequent need for early transplantation (Narayanaswamy et al, 2007).
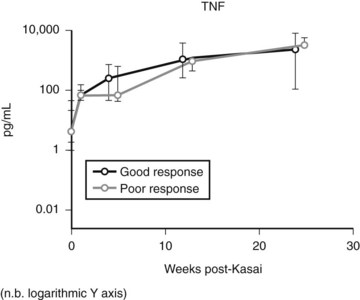
Resident (Kupffer cells) or systemic/recruited macrophages and monocytes seem to play a dual role in BA, as both the presenters of antigenic material in the first place and as the initiating driver for fibrosis in the development of chronic liver disease. Tracy and colleagues (1996) first showed increases in resident macrophages (CD68+) with marked expression of the lipopolysaccharide receptor CD14+. Increased levels of both CD68+ cells and circulating markers of CD68+, TNF-α, and IL-18, have been shown to predict outcome post-Kasai (Kobayashi et al, 1997; Narayanaswamy et al, 2007).
Viral Hypothesis of Biliary Atresia
One suggested cause of BA is infection with hepatotropic viruses, such as rotavirus, reovirus, and cytomegalovirus, either as an indirect trigger of an abnormal immune-mediated reaction or as an actual pathologic agent. The evidence is largely based on animal models in which intraperitoneal innoculation on the first day of life is able to mimic postnatal biliary inflammation (Riepenhoff-Talty et al, 1993; Petersen et al, 1998). Subsequent translation into humans has been more problematic. Serologic evidence of viremia is contradictory (Morecki et al, 1982; Tarr et al, 1996), and isolation of both viral RNA and DNA have (Riepenhoff-Talty et al, 1996; Tyler et al, 1998; Fischler et al, 1999) and have not (Brown et al, 1988; Steele et al, 1995; Bobo et al, 1997; Jevon & Dimmick, 1999) been identified by PCR techniques.
The most recent study is one from Hannover, Germany, in which liver biopsies from 74 infants obtained at the time of portoenterostomy were tested for a panel of DNA and RNA hepatotropic viruses (Rauschenfels et al, 2009). One or more viruses was detected in about one third of infants, and the rate of detection increased with infant age, which suggested that observed viral infection was more likely to be a secondary finding rather than a specific cause.
One problem with the viral hypothesis is the lack of an explanation as to why such ubiquitous organisms only damage the biliary tree in such a small fraction of infants and children. It may be that such infants are in some way susceptible to cholangiocyte damage and require a (viral) trigger to set off an immune-mediated destructive process (Mack, 2007).
RNA gene expression studies in rotavirus-induced murine models have shown upregulation of many proinflammatory genes, among them the IFN-γ inducers IRF7 and IRF9 (Carvalho et al, 2005). Similar gene-expression profiling in human BA samples has also shown overexpression of immune regulatory genes and differences between isolated and syndromic forms (Bezerra et al, 2002; Zhang et al, 2004).
A number of studies have shown different but specific elements of genetic variation between patients with isolated BA and unaffected control subjects. Thus, susceptibility may be imparted by polymorphisms in a number of relevant genes. Examples of this include vascular endothelial growth factor (VEGFA; Lee et al, 2010), macrophage migration inhibitory factor (MIF, Arikan et al, 2006), and intercellular adhesion molecules (ICAMs; Arikan et al, 2008).
Most recently, maternal microchimerism, a novel mechanism of immune damage, has been suggested based on the observation that male infants with BA have a threefold increase in cells of maternal origin in their livers (Hayashida et al, 2007). These were later shown to be maternal-origin chimeric CD8+ T cells and CD45 NK cells capable of initiating immune cholangiolar damage (Muraji et al, 2008).
Clinical Features
The cardinal features of BA are conjugated jaundice persisting beyond 14 days of age, pale and unpigmented stools, and dark urine in an otherwise healthy, term neonate. Liver fibrosis and cirrhosis are later developments, even in infants with intrauterine BA evident at birth (Makin et al, 2009); therefore ascites and marked hepatosplenomegaly are rarely seen before 3 months. Fat malabsorption occurs by stealth, and these infants will fall behind in their growth (Livesey et al, 2009). Fat malabsorption can also cause fat-soluble vitamin K deficiency, and some infants (<3%) will be seen with a possibly catastrophic bleeding tendency and a raised international normalized ratio (INR).
A proportion of infants (approximately 5%) will be seen initially with an abnormal antenatal ultrasound (US) scan (Caponcelli et al, 2008). These will be shown to have cystic BA, with the “cyst” evident from at least 20 weeks’ gestation. This has no untoward effects during pregnancy but clearly needs thorough investigation postnatally. The differential diagnosis for these infants, if the cyst is actually recognized as of hepatobiliary origin, is that of a cystic choledochal malformation, better known as a choledochal cyst (Hinds et al, 2004).
Diagnostic Workup
US examination is a key part of the workup in order to exclude other possible surgical diagnoses (choledochal malformation, inspissated bile syndrome), and evidence of at least intrahepatic bile duct dilation should be visible (Davenport et al, 2003). Although suggestive and supportive, US is uncommonly diagnostic of BA and typically shows a shrunken, atrophic gallbladder with no evidence of filling between feeds. Some US signs do appear more reliable, such as the “triangular cord sign” (Park et al, 1997). This suggests a sonographic appearance of the proximal solid biliary remnant in front of the bifurcation of the portal vein, and advocates quote accuracy rates of greater than 80% (Humphrey & Stringer, 2007).
Direct cholangiography is becoming more prevalent in larger centers with direct access to endoscopic retrograde cholangiopancreatography (ERCP) (Shanmugam et al, 2009; Petersen et al, 2009
Related posts:
 Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
Preoperative and postoperative nutrition in hepatobiliary surgery
Medical management of bleeding varices: Primary and secondary prophylaxis
 Portal hypertension in children
Portal hypertension in children
 Liver transplantation for cholangiocarcinoma and other neoplastic diseases
Liver transplantation for cholangiocarcinoma and other neoplastic diseases
 Distal splenorenal shunt
Distal splenorenal shunt
 Interventional endoscopy: Technical aspects
Interventional endoscopy: Technical aspects
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


