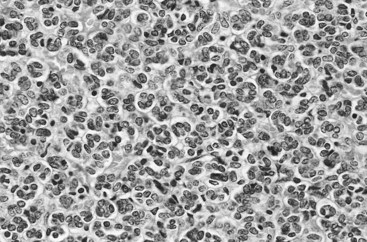
Vitaly Margulis, MD, Surena F. Matin, MD, Christopher G. Wood, MD Benign renal neoplasms constitute a rather large and heterogeneous group of renal lesions that can be found in the kidney. These include the simple renal cyst, selected complex renal cysts, cortical and metanephric adenomas, angiomyolipoma, oncocytoma, the rarer cystic nephroma, mixed epithelial/stromal tumor, and leiomyoma, as well as other, even more esoteric tumor types. Management approaches to these lesions can vary widely, from no management for the simple renal cyst to selective embolization for larger angiomyolipomas and surgical extirpation for solid renal masses when the differential diagnosis includes renal cell carcinoma (RCC). With the increased use of cross-sectional abdominal imaging for renal-specific as well as other nonspecific complaints, it is expected that the identification of both benign and malignant renal tumors will continue to increase in the coming years (Patard, 2009). Moreover, with the increase use of, and refinements to, renal mass biopsy, the management of both benign and malignant renal neoplasms is still evolving in a way that the indications for intervention, and the type of intervention, may change significantly in the coming years (Lane et al, 2008; Campbell et al, 2009). Now, with advanced multidimensional imaging techniques, as well as minimally invasive, nephron-sparing surgical approaches, percutaneous ablative approaches, and the concept of active surveillance in the armamentarium of the practicing urologist, management of all renal lesions, including those that are presumptively benign in etiology, continues to evolve (Raj et al, 2007; Benway and Bhayani, 2009; Murphy et al, 2009). At present, however, the urologist is left primarily with imaging studies such as ultrasonography, computed tomography (CT), or magnetic resonance imaging (MRI) to assess whether a renal lesion is benign or malignant before therapeutic decisions are made. And unless the mass has clear radiographic features that suggest a benign etiology, such as clear evidence of fat seen with most angiomyolipomas or the smooth walls and lack of enhancement of a simple or minimally complex cyst, the vast majority of benign renal lesions are diagnosed only after definitive therapy such as surgical intervention has been implemented. There are several clinical features that have been linked to an increased likelihood of a renal mass having a benign etiology, including smaller size, female sex, and older age, but none of these factors can be relied on to avoid intervention if a specific pretherapy diagnosis cannot be made (Kutikov et al, 2006; Snyder et al, 2006; Glassman et al, 2007; Lane et al, 2007; Beisland et al, 2009; Murphy et al, 2009). In much the same way as the molecular biology of RCC was elucidated through the study of familial genetic syndromes such as von Hippel-Lindau disease, the molecular basis for cyst formation has been further elucidated through the genetic analysis of familial renal cystic syndromes, such as autosomal dominant polycystic kidney disease (ADPKD). Through these studies researchers have identified that loss of specific genes, such as PKD1 and/or PKD2 (polycystins) lead to cyst formation in patients with ADPKD. Polycystin-1 and polycystin-2 form a complex that represents a critical ion channel in the kidney, the loss of which results in cyst formation through the loss of intracellular calcium dysregulation and dysfunction of the primary renal cilia, a poorly understood organelle that is in part responsible for normal renal development (Pei, 2003; Weimbs, 2007; Ibraghimov-Beskrovnaya and Bukanov, 2008). Whether these genetic changes are common to sporadic forming “benign” renal cystic disease remains to be proven, but many of the phenotypic and genetic changes noted in the kidneys of patients with familial cystic disease syndromes have been identified in sporadic cystic disease (Qian et al, 1996; Pei, 2001). Perhaps the best description of the natural history of sporadic renal cysts can be found in the recently updated study by Terada and associates (2008). In this study of 61 patients with simple renal cysts and a mean of 10 years of follow-up, the authors noted that the cysts increased in both size and number over time. The average increase in size of the cysts was 1.9 mm per year, but the authors also noted that the rate of size increase decreased with age. Interestingly, two cysts developed renal neoplasms during the course of the study and no differences were noted in the clinical characteristics of the cysts that developed neoplasms from those that did not. Regarding the risk factors for the development of cysts, increasing age, male gender, presence of hypertension, and the presence of renal insufficiency were all associated with the development of sporadic renal cysts (Terada et al, 2004). Renal cysts remain the most common benign renal lesions, representing more than 70% of asymptomatic renal masses. They can be solitary or multiple and unilateral or bilateral (Terada et al, 2002). In addition to sporadic cystic disease of the kidney, and the cysts that occur with familial syndromes such as ADPKD and autosomal recessive polycystic disease, cysts are also known to occur in association with end-stage renal disease in patients on dialysis (Bisceglia et al, 2006). There appears to be a higher incidence of RCC associated with the development of acquired renal cystic disease, much like that seen with von Hippel-Lindau disease and tuberous sclerosis, such that the pathogenesis of these cystic lesions may be quite different from that seen with sporadic simple or minimally complex cysts (Truong et al, 2003). Renal cystic lesions can be imaged through a variety of radiographic techniques, including ultrasonography, CT, as well as MRI. On ultrasonography, simple renal cysts have a smooth wall, are fluid filled with no internal echoes, and have evidence of posterior wall enhancement. Evidence of internal echoes, calcifications or nodularity in the wall, or internal septa on ultrasonography suggest a more complex cyst that might be worthy of further imaging that includes intravenous administration of a contrast agent (Quaia et al, 2008; Eknoyan, 2009). The Bosniak classification for renal cystic lesions, as reviewed in Table 51–1, is perhaps the most useful and widely employed method for characterizing renal cystic lesions and for assessing the likelihood of the presence of a concomitant malignancy within the cyst (Israel and Bosniak, 2005; Warren and McFarlane, 2005). In general, Bosniak class I, II, and IIF cysts are likely to represent benign lesions, thus requiring either no therapy or just continued radiographic follow-up, in the case of class IIF lesions (Fig. 51–1). These recommendations are based on a number of series published in the literature by Bosniak and others that include both radiographic as well as pathologic follow-up (Warren and McFarlane, 2005). Because of the higher risk of malignancy associated with Bosniak class III and IV lesions, therapy is recommended (Fig. 51–2). The definitive therapy would be surgical excision, although ablative therapies such as cryotherapy or radiofrequency ablation of cystic masses have been reported (Raman et al, 2009) (see Chapter 49). The overwhelming majority of simple or minimally complex cysts require no further follow-up or therapy once diagnosed (Eknoyan, 2009). Rarely, benign cystic lesions of the kidney can grow to such a large size that they can cause pain or other symptomatology, including hypertension (Porpiglia et al, 2009; Zerem et al, 2009). Symptoms can also occur as a consequence of hemorrhage within the cyst or spontaneous/traumatic cyst rupture (Hughes et al, 1995; Rainio et al, 2006; Ishikawa et al, 2008; Vaidyanathan et al, 2008). A variety of therapeutic interventions have been described for benign symptomatic cystic lesions of the kidney. These include aspiration, surgical resection, cyst decortications, as well as sclerotherapy with a variety of different agents (Cho et al, 2008; Ham et al, 2008; Baysal and Soylu, 2009; Canguven et al, 2009; Choi et al, 2009; Porpiglia et al, 2009). Although none of these approaches seems to be better than the others described, it is noted that with aspiration and sclerotherapy there is a higher incidence of cyst recurrence and multiple treatments may be needed to satisfactorily ablate the cystic lesion. A word of caution is warranted regarding the treatment of peripelvic cysts: given their proximity to the vital structures of the kidney, including the renal vessels and collecting system, laparoscopic, rather than percutaneous, approaches may be associated with a higher safety margin and better efficacy (Okumura et al, 2003; Camargo et al, 2005). The designation and management of purportedly benign renal cortical adenomas remains a subject of controversy in the urologic literature. These lesions are small, solid cortical renal lesions that are thought to have a benign course (Renshaw, 2002). Histologically, renal adenomas appear as small, well-circumscribed lesions characterized by uniform basophilic or eosinophilic cells with benign-appearing nuclear and cellular features, arranged as tubulopapillary or purely papillary growth. The incidence of renal adenomas increases with age and male sex, and these tumors also have been associated with acquired renal cystic disease that results in end-stage renal failure (Leroy et al, 2001; Denton et al, 2002; Snyder et al, 2006; Ferda et al, 2007). The incidence of renal adenomas in autopsy series ranges from 7% to 23%, although antemortem pathologic diagnosis of renal adenoma is much less common, in part owing to the pathologist’s concern that there are no reliable histopathologic, ultrastructural, or immunohistochemical criteria to distinguish benign from malignant lesions of the kidney (Licht, 1995). In fact, in a more recent study in which immunohistochemical analyses were used to further characterize renal adenomas, it was suggested that these lesions may be linked to the development of papillary RCC and represent a biologic link and continuum as a premalignant precursor (Wang et al, 2007). In this study, researchers examined 542 nephrectomy specimens obtained over 8 years. Seven percent of these demonstrated evidence of renal adenoma; and of these, 47% were associated with a concomitant papillary RCC. Adenomas that arose in the setting of papillary RCC tended to be multiple (61%). Eighty-two percent of adenomas found in the study had a similar immunohistochemistry staining profile as papillary RCC, staining positive for α-methyl-coenzyme A racemase. In other studies, renal adenomas shared similar cytogenetic profiles to papillary RCC, such as trisomy of chromosomes 7 and 17, thus suggesting a biologic link between the two neoplasms (Brunelli et al, 2003a, 2003b). The overwhelming majority of renal adenomas remain asymptomatic, are undetectable radiographically due to their small size (<1 cm), and require no further therapy. Size of the neoplasm has historically been utilized to differentiate renal adenoma from more malignant neoplasms of the kidney. Thoenes and colleagues (1986) provided a reassessment of the histologic classification of renal tumors and defined renal adenoma as a tumor with nuclear grade I and a diameter of at least 1 cm. For many years, a “3-cm rule” was pervasive in the urologic literature, dating back to the original autopsy studies by Bell (1938) when he noted that only 1 of 38 renal cortical tumors had associated metastases whereas 70 of 106 tumors larger than 3 cm were associated with metastases. The diagnosis of renal adenoma remains controversial; many believe that all solid renal epithelium-derived masses are potentially malignant and therefore should undergo treatment (Renshaw, 2002). In 1995 Davis and colleagues reported 50 cases of an unusual and novel renal mass lesion with distinctive histologic features and a benign clinical course despite occasional symptomatic presentation and large tumor size. In this series, mean tumor size was 5.5 cm (up to 15 cm) and nearly half of the patients presented with flank pain, gross hematuria, or a palpable mass. Six additional patients presented with polycythemia, and hypercalcemia has also been reported in association with this tumor type, which was designated metanephric adenoma (Davis et al, 1995; Mahoney et al, 1997; Kuroda et al, 2003b). Metanephric adenoma was officially accepted as a primary benign renal tumor based on consensus from the Heidelberg classification (Kovacs et al, 1997). A higher male-to-female ratio was initially reported in the series of Davis and colleagues (1995), but it is likely that there is actually a female predominance as has been reported in other series (Jones et al, 1995; Snyder et al, 2006; Bastide et al, 2009). Incidental presentation is most common, and peak incidence is in the fifth decade of life (Renshaw, 2002). Polycythemia can be seen in 10% of patients and appears to be due to the production of erythropoietin and other cytokines by the tumor (Yoshioka et al, 2007; Bastide et al, 2009). Radiographically these tumors can have peripheral or central calcifications and may be hypovascular on contrast-enhanced CT and hyperechoic on ultrasonography (Bastide et al, 2009). On microscopic examination these tumors consist of very small, often highly basophilic epithelial cells that form small acini and occasionally tubular or papillary structures within a predominantly acellular stroma (Fig. 51–3). Davis and colleagues (Renshaw, 2002) argued that metanephric adenoma might be histologically related to epithelial Wilms tumor because they believed that it exhibited histologic similarities to the metanephric, hamartomatous elements of nephroblastomatosis. Along these lines, it is interesting to note that many of these tumors exhibit evidence of regression in the form of scarring or calcification. In addition, Muir and colleagues (2001) have shown positive staining for the Wilms tumor protein WT1 and an immunohistochemical staining profile that suggests a histogenetic relationship to Wilms tumor. An alternative theory for the origin of metanephric adenoma was proposed by Brown and associates (1997), who found gain of chromosomes 7 and 17 by fluorescent in-situ hybridization in 8 of 11 of these tumors. These findings suggest a clonal neoplastic disorder potentially related to papillary RCC, but others have argued that this series may have been contaminated by inclusion of some cases of papillary RCC, which can be difficult to differentiate from metanephric adenoma (Brunelli et al, 2003a). Arroyo and colleagues (2001) showed that metanephric adenomas are part of a spectrum of related tumors, bridging metanephric stromal tumors, metanephric adenofibromas, and Wilms tumor. Pesti and colleagues (2001) have described a putative tumor suppressor gene for metanephric adenoma at chromosome 2p13. Various histologic stains have been evaluated to help distinguish metanephric adenoma from other renal neoplasms. The Wilms tumor marker WT1 is frequently expressed in metanephric adenoma (Muir et al, 2001; Bosco et al, 2007). α-Methylacyl-CoA racemase (AMACR) is poorly expressed in metanephric adenoma but highly expressed in papillary RCC (Olgac et al, 2006), whereas S-100 protein expression is very high in metanephric adenoma, weak in Wilms tumor, and absent in papillary RCC (Azabdaftari et al, 2008). Skinnider and colleagues (2005) demonstrated the potential utility of an expression panel to help differentiate papillary RCC and metanephric adenoma using a panel of cytokeratins 7, 8, 18, and 19 and vimentin. Using these various markers appears to have improved the diagnostic yield of percutaneous biopsy and fine-needle aspiration (Bosco et al, 2007; Patel et al, 2009), but the index of initial suspicion needs to be high for biopsy to be considered. Figure 51–3 Classic metanephric adenoma with small, intensely basophilic cells arranged in an acinar pattern. Only one case of metastasis has been described in association with classic metanephric adenoma into a regional lymph node, and death related to this entity has not been reported (Renshaw, 2002). However, Picken and colleagues (2001) have described malignant stromal elements associated with a metanephric neoplasm of the kidney in a 21-year-old woman who died of progressive cancer, and they have proposed that there may be a spectrum of metanephric tumors that includes rare, aggressive variants. Atypical histologic features and multifocality in childhood have also been reported (Jain et al, 2007; Kohashi et al, 2009). Given the rarity of this tumor and the lack of highly predictive clinical or radiographic criteria, metanephric adenoma remains primarily a pathologic diagnosis. If radiographic findings raise the index of suspicion, then percutaneous core biopsy with fine-needle aspiration may prove helpful in establishing a diagnosis for nephron-sparing treatment or observation, but most patients will require surgical excision because of concern for malignancy (Fig. 51–4). Key Points: Metanephric Adenoma Renal oncocytoma is the most common benign tumor that appears as an enhancing renal mass on cross-sectional imaging and is presumed to be RCC until surgical excision, representing one of the ultimate challenges in preoperative diagnosis for the urologist. It accounts for 3% to 7% of kidney tumors (Morra and Das, 1993). Oncocytoma initially became accepted as a distinct entity after a report of 13 cases in 1976 by Klein and Valensi. Multiple additional reports since that time, including more recent genotyping studies, confirm it to be a benign histology with a distinct cell of origin and genetic abnormalities (Lieber et al, 1987; Davis et al, 1991; Licht et al, 1993; Perez-Ordonez et al, 1997; Amin et al, 1997; Dechet et al, 1999; Chao et al, 2002; Kuroda et al, 2003a). Cases of metastatic disease have been reported, yet these are considered exceptionally rare and may represent cases of malignant degeneration or pseudometastases (Paner et al, 2005; Oxley et al, 2007). Grossly these tumors are mahogany or tan, homogeneous, and well circumscribed with a pseudocapsule and typically a central stellate scar (Fig. 51–5). Microscopically the cells are round or polygonal and arranged in a nested growth pattern. The cells are uniform and highly eosinophilic, owing to an abundance of mitochondria (Renshaw, 2002). In up to one third of cases, hemorrhage, extension into perinephric fat, cellular atypia, prominent nucleoli, and pleomorphism may be seen, yet the clinical behavior in these cases is within what is expected with a benign course (Amin et al, 1997; Perez-Ordonez et al, 1997). The most common genetic abnormality is loss of chromosome 1p (Lindgren et al, 2004; Paner et al, 2007). Other common cytogenetic findings include loss of the Y chromosome and chromosome 14q and rearrangements of 11q13 (Schwerdtle et al, 1997; Chao et al, 2002; Polascik et al, 2002; Lindgren et al, 2004). The chromosomal abnormalities typically seen in RCC are not seen in renal oncocytomas, further reinforcing the concept that these tumors are genotypically distinct from RCC (Minor et al, 2003). However, histologically, the greatest dilemma arises from distinguishing chromophobe and clear cell RCC with eosinophilic characteristics from oncocytoma. Hale colloidal iron staining is the classic differentiating marker for oncocytoma, but it can have nonspecific staining and be difficult to interpret (Leroy et al, 2000). Cytokeratin profiles are helpful in distinguishing these histologic findings (Skinnider et al, 2005; Adley et al, 2006). Expression of cytokeratin-7 is seen in 66% of chromophobe RCC and only 5% of oncocytomas, and parvalbumin is expressed in 100% of chromophobe RCC and 47% of oncocytomas (Leroy et al, 2000; Adley et al, 2006). A variety of other markers have been recently described to differentiate oncocytoma from RCC, particularly chromophobe RCC, but the clinical utility for most of these has not yet been fully developed. These include Pax-2, expressed in metanephric tissues and vital for renal tubule development; pattern of claudin-7 and 8 expression, tight junction proteins expressed in distal nephron epithelium; a vimentin expression pattern; c-KIT expression; and S-100 (Pan et al, 2004; Lin et al, 2006; Hes et al, 2007; Rocca et al, 2007; Lechpammer et al, 2008; Gupta et al, 2009; Osunkoya et al, 2009). An “optimal” panel for distinguishing between chromophobe and clear cell RCC and oncocytoma was recommended by Liu and colleagues in 2007, consisting of a combination of three markers (vimentin, glutathione-S-transferase-α, and epithelial cell adhesion molecule). These investigators achieved 100% sensitivity and 100% specificity for the differential diagnosis of chromophobe carcinoma, oncocytoma, and clear cell carcinoma (Liu et al, 2007). The feasibility of molecular signatures and gene profiling is being evaluated as potentially useful tests for the accurate diagnosis of tumors such as oncocytoma (Schuetz et al, 2005; Yang et al, 2006), but such studies are not yet a clinical reality. As noted previously there are several distinct similarities to chromophobe RCC, which also is derived from the distal renal tubules. Chromophobe RCC, particularly the eosinophilic variant, and oncocytoma are histologically similar and their distinction often requires additional pathologic testing (Weiss et al, 1995; Renshaw, 2002). The commonality of these two tumors is further evidenced in patients with Birt-Hogg-Dubé syndrome, in whom both oncocytomas and chromophobe RCC develop, in addition to cutaneous fibrofolliculomas and spontaneous pneumothoraces (Pavlovich et al, 2005; Toro et al, 2008). Some of the renal tumors display a histology between these two tumors, prompting some to speculate that chromophobe RCC and oncocytoma represent points in a spectrum of neoplasia (Chao et al, 2002; Linehan, 2003; Pavlovich et al, 2005; Toro et al, 2008). The greatest clinical dilemma remains the inability to confidently differentiate between renal oncocytoma and RCC on clinical or radiographic testing. Both have a similar age at presentation, have a male-to-female predominance, and are similarly sized at presentation. Although oncocytomas were more likely to be asymptomatic at presentation, most RCC are also diagnosed incidentally in the current era, eliminating this clinical scenario as a differentiator (Davis et al, 1991; Licht et al, 1993; Lieber, 1993; Amin et al, 1997; Perez-Ordonez et al, 1997; Dechet et al, 1999). The typical spoke-wheel pattern seen on angiography or the stellate scar on cross-sectional imaging may bring up the question of a renal oncocytoma, but these findings have a poor predictive value by themselves (Davidson et al, 1993; Licht et al, 1993; Licht, 1995). On MRI, oncocytomas may have distinct T1 and T2 signal patterns that can be suggestive but these are not definitive findings (Harmon et al, 1996). T2-weighted images on MRI do not differentiate oncocytoma from RCC (Dann et al, 2006). For lesions undergoing surveillance, the growth rates of RCC and oncocytoma are similar, so growth kinetics also do not help differentiate these tumors either (Chawla et al, 2006; Crispen and Uzzo, 2007; Siu et al, 2007). In 4% to 13% of cases, tumors are multicentric, are bilateral, or have a metachronous presentation (Lieber et al, 1987; Davis et al, 1991; Licht et al, 1993; Amin et al, 1997; Perez-Ordonez et al, 1997; Dechet et al, 1999; Tickoo et al, 1999; Minor et al, 2003). Familial renal oncocytomatosis was initially described (Weirich et al, 1998) in five families in which it presented at a young age as multicentric, bilateral and recurrent oncocytomas. Nonfamilial forms of bilateral multifocal oncocytomas resembling oncocytomatosis can also occur (Fig. 51–6). A recent cytogenetic evaluation of a patient with apparently sporadic oncocytomatosis and hybrid tumors showed different chromosomal losses than the Birt-Hogg-Dubé syndrome (Al-Saleem et al, 2004). Recent clinical studies highlight the differing incidence of oncocytoma based on age and gender. Cao and colleagues (2005) and Skolarus and colleagues (2008) showed an increasing incidence of oncocytoma in older patients presenting with a small incidentally discovered renal mass. Two different reports highlight that younger females are nearly twice as likely as their male counterparts to have a benign tumor, which includes oncocytoma and angiomyolipoma but is probably largely driven by the higher rates of angiomyolipoma in women (Cao et al, 2005; Snyder et al, 2006). Historically fine-needle aspiration or core biopsy was compromised by high false-negative or nondiagnostic specimens, given the difficulty in differentiating oncocytoma with RCC subtypes (Weiss et al, 1995; Campbell et al, 1997). However, the diagnostic accuracy of percutaneous biopsy has markedly improved, particularly when a core biopsy is done in addition to a fine-needle aspiration and bolstered with the use of immunostains (Liu and Fanning, 2001; Barocas et al, 2006; Lebret et al, 2007; Volpe et al, 2007; Kummerlin et al, 2008; Schmidbauer et al, 2008), prompting some investigators to revisit the role of the biopsy in the management of some patients with an incidental renal tumor (Shah et al, 2005; Lebret et al, 2007; Volpe et al, 2007). When multiple tumors are present, one must consider the possibility of RCC coexisting with oncocytoma, which in some series has been shown to be as high as 32% (Davis et al, 1991; Licht et al, 1993; Licht, 1995; Gudbjartsson et al, 2005). Consideration of biopsy in these cases must be thoughtfully considered to provide a sampling of all sites of disease that are of concern. Treatment options for a known oncocytoma range from observation to thermal ablation, laparoscopic or open partial nephrectomy, and even radical nephrectomy depending on the clinical scenario and uncertainty regarding the diagnosis (Licht, 1995; Romis et al, 2004; Gudbjartsson et al, 2005; Crispen and Uzzo, 2007). If oncocytoma is highly suspected and surgery is indicated, a nephron-sparing approach is preferred given the benign nature of these lesions and the very low probability of recurrence (Licht, 1995; Romis et al, 2004; Gudbjartsson et al, 2005). Frozen section analysis is usually not sensitive enough to differentiate the eosinophilic appearance of oncocytomas with eosinophilic RCCs and should not be used to guide surgical strategy. Thermal ablation, although sometimes reported as a treatment option, commits the patient to long-term radiographic surveillance, given the lower success rates of these procedures and the unknown long-term outcomes, tests that would be considered unnecessary should surgical excision be performed. In most cases the treatment options are isolated to observation, particularly for the older or sicker patient, and surgical resection, particularly for the younger healthier patient. Angiomyolipoma accounts for less than 10% of renal tumors, with autopsy series and ultrasound-screened populations showing a 0.3% and 0.13% incidence in the general population (Eble, 1998). It is a benign neoplasm consisting of thick-walled aneurysmal vessels, smooth muscle, and varying levels of mature adipose tissue (Tamboli et al, 2000; Nelson et al, 2002; Bissler et al, 2004). Initially considered to be a form of hamartoma or choristoma, recent evidence suggests a neoplastic origin with evidence of a monoclonal, rather than polyclonal, source (Green et al, 1996; Sepp et al, 1996; Kattar et al, 1999). Angiomyolipoma is now considered to be of neural crest origin, possibly derived from perivascular epithelioid cells (PEC). Originally described in 1911 by Fischer, its current name was given by Morgan in 1951 (Eble, 1998). The tumor strongly expresses estrogen receptor-β as well as androgen receptor, is predominantly found in females, and is rare before puberty, suggesting a potential hormonal influence (Boorjian et al, 2008). The typical sporadic presentation is of a middle-aged woman with a single asymptomatic tumor. Sporadic angiomyolipomas appear to have a slow growth rate and are usually detected incidentally (Seyam et al, 2008). Angiomyolipoma is the most common renal neoplasm associated with spontaneous perirenal hemorrhage, closely followed by RCC (Zhang et al, 2003). Skolarus and colleagues (2008) suggested that there may be a decreased incidence of sporadic angiomyolipoma with increasing age. These tumors are most often sporadic but can also be associated with the autosomal dominant tuberous sclerosis complex (TSC). Twenty to 30 percent of angiomyolipomas are in patients with TSC, and approximately 50% of patients with TSC develop angiomyolipomas (Eble, 1998; Neumann et al, 1998; Tamboli et al, 2000; Lendvay et al, 2003; Minor et al, 2003). TSC-associated angiomyolipomas typically present at a younger age (mean age 30 years); there is less female-to-male predominance (2 : 1) with multiple, bilateral, and symptomatic tumors (Eble, 1998
Renal Cysts
Renal Cortical Adenoma
Metanephric Adenoma
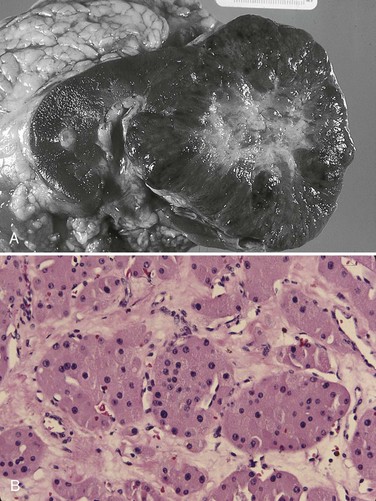
Oncocytoma
Angiomyolipoma
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Prostate Cancer Tumor Markers
Prostate Cancer Tumor Markers
 Core Principles of Perioperative Management in Children
Core Principles of Perioperative Management in Children
 Neuropathic Dysfunction of the Lower Urinary Tract
Neuropathic Dysfunction of the Lower Urinary Tract
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Benign Renal Tumors

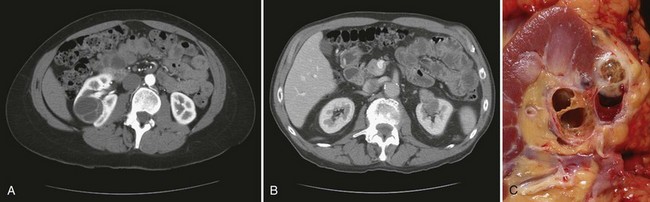
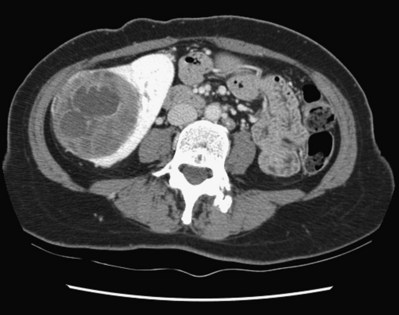
• Metanephric adenoma is a recently described, rare benign mass that radiographically may be indistinguishable from RCC.