Stuart M. Flechner, MD, FACS, James H. Finke, PhD, Robert L. Fairchild, PhD Immune mechanisms rely upon the interactions between diverse cell populations, which at any point in time are in various stages of differentiation. In contemporary terms immunity can be divided into two separate responses. Cell-mediated immune responses describe those primarily directed by cell-to-cell contact, and humoral immune responses describe those primarily due to the production of antibody (immunoglobulin). In fact, most responses involve both cellular and humoral events, which are mediated by several different types of immune competent cells. Although the small lymphocyte is the backbone of the immune response, several other bone marrow–derived cells are needed for a mature immune response. These cells “talk” to each other and their surroundings through receptors that they express on their surface, and through cytokines, which are small peptides that they secrete locally (Clark and Ledbetter, 1994). The receptors are usually glycoproteins that extend from the cell surface as single chains or double chains called dimers, which are often designated alpha and beta. A nomenclature has developed to describe these markers, at international workshops, comparing several monoclonal antibodies from different laboratories. When a cluster of monoclonals are found to react with the same surface polypeptide, it is designated with a CD number (cluster of differentiation). The number of CD specificities on leukocytes is now in the hundreds, and Table 17–1 provides the ones more commonly associated with immune responses. A cell surface receptor bound by its ligand will often trigger downstream intracellular events, thereby activating the cell to either produce specific cytokines, express other surface markers required for immune interactions, or enter cell cycle progression and proliferation. Many of the small peptide cytokines that directly influence immune responses will be described further in this chapter, and are represented in Table 17–2. Table 17–1 Some of the Major Cell Surface CD (Cluster of Differentiation) Markers
| CD NO. | CELLULAR ASSOCIATION | MEMBRANE COMPONENT/LIGAND |
|---|---|---|
| CD2 | T cells, NK cells | Receptor for LFA3 (CD58), sheep RBC rosettes |
| CD3 | T cells | Component of T-cell receptor, signal transduction |
| CD4 | Helper T cells | Receptor for MHC class II and HIV virus |
| CD5 | T cells, thymocytes | Signaling molecule, binds CD72 |
| CD8 | Cytotoxic T cells | Receptor for MHC class I |
| CD11a | Leukocytes | Adhesion binds ICAMs (CD50, 54, and 102) |
| CD16 | PMN, NK, macrophages | Fcγ receptor, phagocytosis, and ADCC |
| CD18 | All leukocytes | Integrin beta chain |
| CD19 | B cells | Pan–B-cell marker |
| CD20 | B cells | Role in B-cell activation |
| CD25 | Activated T and B cells, macrophages | IL-2 receptor, also called Tac |
| CD28 | T cells | Costimulator ligand for B7.1 (CD80) and B7.2 (CD86) |
| CD40 | B cells, APCs | Receptor for CD40 ligand (CD154), B-cell costimulator |
| CD44 | Leukocytes | Proteoglycan binding receptor, |
| CD45 | Leukocytes | Leukocyte common antigen, tyrosine phosphatase |
| CD45RO | Activated and memory T and B cell subset, monocytes, macrophages | Isoform of CD45, CD45RO memory cells; high MW |
| CD45RA naive cells; low MW | ||
| CD52 | T and B cells, some monocytes, macrophages | CAMPATH-1, unknown function |
| CD54 | Dendritic, macrophages, B cells | Leukocyte adhesion, also called ICAM-1 |
| CD55 | Most cells | Decay accelerating factor (DAF), binds C3b |
| CD56 | NK, few T cells | Adhesion |
| CD58 | Broad | Binds CD2 (called LFA-3) |
| CD62L | B and T cells, monocytes, PMN | L-Selectin, endothelial adhesion, homing of naive T cells to lymph nodes |
| CD69 | Activated leukocytes | Activation inducer molecule (AIM), signal transduction |
| CD71 | Erythroid cells | Transferrin receptor, iron uptake |
| CD80 | Dendritic, macrophages, B cells | Ligand for CD28, also B7.1, T-cell costimulator |
| CD86 | Dendritic, macrophages, B cells | Ligand for CD28 and CD152 (CTLA-4), also B7.2 |
| T-cell costimulator | ||
| CD152 | Activated T cells | Also called CTLA-4; negative regulation of costimulation; binds CD80 (B7.1) and CD86 (B7.2) |
| CD154 | Activated CD4+ T cells | CD40 ligand, activates B cells |
AM, adhesion molecules; MW, molecular weight; NK cells, natural killer cells; PMN, polymorphonuclear leukocytes; RBC, red blood cells.
Table 17–2 Representative Cytokines
| CYTOKINE | CELLULAR SOURCE | MAJOR ACTIVITIES |
|---|---|---|
| IL-1 | MØ, EC, keratinocytes | Upregulation of AM expression, stimulation of CK production, activation of MØ and T cells |
| IL-2 | T cells | Activation of NK cells, T-cell growth factor |
| IL-4 | Type 2 CD4+ and CD8+ T cells | T-cell growth factor, B-cell growth factor, Ig class switching to IgE |
| IL-5 | Type 2 CD4+ and CD8+ T cells | Stimulation of eosinophil development |
| IL-6 | Type 2 CD4+ T cells, MØ | Stimulation of acute phase proteins, activation of lymphocytes |
| IL-7 | Stromal cells bone marrow | Stimulates survival and expansion of immature T and B cells |
| IL-10 | Th2 and Th3 T cells, MØ | Immunosuppression |
| IL-12 | MØ, DC | Stimulation of IFN-γ production |
| IL-13 | Type 2 CD4+ T cells | Allergic inflammation, upregulation of VCAM-1 expression |
| IL-15 | Fibroblasts, EC, DC | CD8+ T-cell growth factor |
| IL-17 | CD4+ T Cells | Promotes inflammatory responses; may trigger autoimmunity |
| TNF-α | T cells, NK cells, MØ | Upregulation of AM expression, stimulation of CK production, inflammation |
| TNF-β | Type 1 CD4+ and CD8+ T cells | MØ activation, cytolysis |
| IFN-γ | Type 1 CD4+ and CD8+ T cells, NK cells | MØ activation, upregulation of AM expression, production of T-cell chemoattractants, inhibition of Th2 cells |
| GM-CSF | T cells, NK cells, B cells, MØ | Stimulation of MØ and granulocyte development |
| TGF-β | T cells, B cells, mast cells, MØ | Immunosuppression |
AM, adhesion molecules; CK, chemokines; DC, dendritic cells; EC, endothelial cells; MØ, macrophages; NK cells, natural killer cells.
Innate Immunity
The innate immune responses are constitutive and do not improve on repeated contact with the same offending agent. An invading microbe first encounters innate immune responses. Innate defense mechanisms represent nonspecific barriers to invaders, which rely primarily on physical barriers, phagocytic cells, natural killer cells, complement, acute phase proteins, lysozyme, the interferons, and defensins. Innate immunity also underlies most inflammatory responses; these are initially triggered by macrophages, polymorphonuclear leukocytes, and mast cells through their germline-encoded innate immune receptors (Janeway and Medzhitov, 2002).
Defensins
Defensins are small cysteine-rich peptides of 18 to 45 amino acids that are found in both vertebrates and invertebrates (Klotman and Chang, 2006). They are active against bacteria, fungi, and both enveloped and nonenveloped viruses. Cells of the immune system contain these peptides to assist in killing phagocytized bacteria—for example, in neutrophilic granulocytes and almost all epithelial cells. Most defensins function by binding to microbial cell membranes, and once embedded, forming porelike membrane defects. These defects allow the efflux of essential ions and nutrients, resulting in microbial death.
Phagocytosis
Phagocytosis is a nonspecific innate response of circulating neutrophils and macrophages that actively ingest microbes and secrete antimicrobial toxins. Phagocytosis also includes reticuloendothelial cells such as Kupffer cells in the liver and bronchial alveolar macrophages. The macrophage advances pseudopodia over the microbe, which is internalized into a phagocytic vacuole that fuses with a lysosomal vacuole. This fusion vacuole results in the degradation of the microbe or particle as digestive enzymes are released. Killing is mediated by reactive oxygen intermediates and hydrogen peroxide (Underhill and Ozinsky, 2002).
Natural Killer Cells
These are large granular lymphocytes that can destroy various nucleated cells including tumors and virus-infected cells. They are an important component of innate immunity and do not require prior contact with antigen, nor are they MHC-restricted. NK cells release cytotoxic factors such as perforin, serine proteases called granzymes, and tumor necrosis factor beta (TNF-β). They can also trigger apoptosis in their targets. Interferons seem to augment their activity. They also participate in antibody-dependent cell-mediated cytotoxicity (ADCC), by the binding of their cell surface Fc receptor (CD16) to the Fc portion of antibody-coated cells. The precise lineage of NK cells is not known. Characterized by the presence of CD16 and CD56, and the absence of CD3, they have IL-2 receptors and respond to exogenous IL-2. Although their T-cell receptor (TCR) genes are not rearranged, they seem to be related to T cells (Yokoyama et al, 2004).
Toll-Like Receptors
Toll receptors were originally identified as transmembrane receptors required for the establishment of polarity in the developing Drosophila embryo, but also were shown to be essential in protecting flies from infection. The mammalian counterpart, a family of 10 structurally similar proteins has been identified and named Toll-like receptors (TLRs 1 to 10). Most tissues express at least one TLR, but phagocytes, in particular, show abundant expression of all known TLRs. The mRNA of TLRs has been found in monocytes and macrophages, dendritic cells, mast cells, B cells, and epithelial cells of the respiratory and intestinal tract. Different TLRs respond to different products of microbes, but all of the TLRs activate similar signaling mechanisms, resulting in cellular responses that are central to innate immunity. The TLRs can be present on the cell surface, where they recognize the extracellular products of microbes, and other TLRs are actually part of intracellular endosomes where microbes are ingested. Once TLRs are engaged, they activate transcriptional factors that signal the expression of genes that encode various enzymes, proteins, and cytokines that focus on the antimicrobial functions of phagocytes and dendritic cells (see Immunity to Infections).
Complement
A major component of innate immunity is the collection of about 20 soluble proteins referred to as complement. Complement, once activated, can directly lyse invading bacteria or viruses, and several of its components are important in the regulation of immune responsiveness (Carroll, 2004). Complement activation results in a cascade phenomenon where the product of one reaction is the enzyme catalyst of the next. Each component is described by the letter “C” and a number, more closely reflecting the sequence of discovery rather than sequence of action. The split components are designated with an a or b. The most critical component is C3, which has a molecular weight of about 195 kD. Once complement proteins and a target substance interact, a C3 convertase is activated, which splits C3 into C3a and C3b fragments (Fig. 17–1). The C3b bound to the target becomes a component of the convertase for C5. The union of C5b with C6-C9 generates the membrane attack complex (MAC), which integrates with the lipid bilayer of the target cell. The MAC produces cell membrane lesions that permit loss of potassium and ingress of salt and water leading to hypotonic lysis (Walport, 2001).
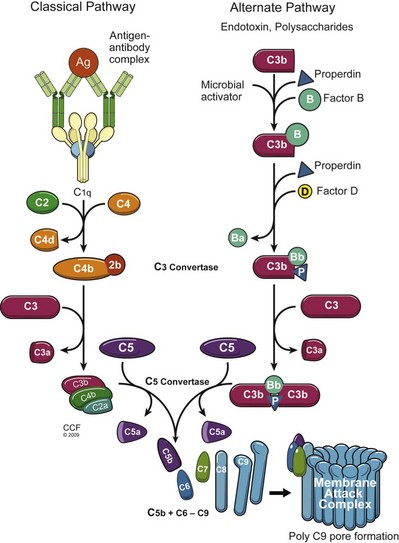
(Reprinted with permission, Cleveland Clinic Center for Medical Art & Photography © 2009-2011. All Rights Reserved.)
Recently, complement activity has become an important target for immune responses to transplanted organs. The human gene for DAF has been transfected into pigs in an attempt to control complement activation during a pig-to-primate xenograft rejection model. In addition, the complement protein split product C4d has been proposed as a reliable marker for antibody-mediated rejection of renal allografts. Antibody-mediated activation of the classical complement pathway leads to the formation of C4d, which covalently binds to the endothelial surface of the kidney peritubular capillaries, thus leaving an imprint of antibody activity that persists for days to weeks (Collins, 1999). C4d-staining in the kidney correlates with the presence of circulating donor-specific antibody in the recipient.
Adaptive Immunity
Lymphoid Organs and the Lymphatic System
Although descriptions of immune responses are often presented as individual events, in vivo they take place within the organized architecture of lymphoid tissue. Bone marrow stem cells differentiate into immunocompetent T and B cells in the primary lymphoid organs, and then colonize the secondary lymphoid tissues where the immune responses take place. The primary lymphoid organs are the thymus, responsible for the selection and education of T cells, and the bone marrow, responsible for the education of B cells. Secondary or peripheral lymphoid tissue includes the spleen, lymph nodes, and unencapsulated tissues lining the alimentary, respiratory, and urogenital tracts (Drayton, 2006). B lymphocytes seed the secondary lymphoid tissues where they form follicles. The spleen filters the blood, the lymph nodes drain local body tissues, and the unencapsulated tissues associated with mucosal surfaces can secrete IgA antibodies. The lymphoid tissues communicate through a network of lymphoid ducts that drain all the viscera and return through the thoracic duct to the circulation (Fig. 17–2). This communication is maintained by populations of recirculating lymphocytes, which can travel from the blood into the spleen, lymph nodes, and peripheral tissues, and return. This traffic, or recirculation, of lymphocytes between the peripheral tissues, lymph nodes, and bloodstream enables the immune system to function as a single organ. Antigen-responsive cells can be recruited to peripheral sites for initial activation, and memory cells can be disseminated to these sites to amplify ongoing responses.
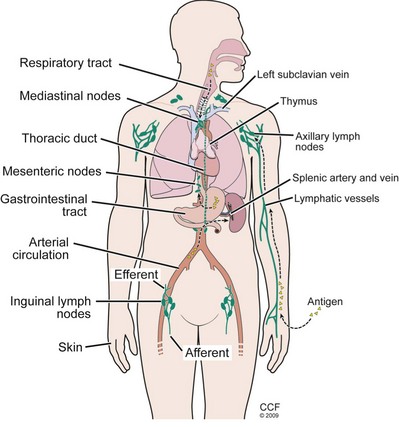
(Reprinted with permission, Cleveland Clinic Center for Medical Art & Photography © 2009-2011. All Rights Reserved.)
The lymph nodes, located along lymphatic channels throughout the body, are the primary nexus where naive immunocompetent cells meet a foreign antigen (Fig. 17–3). Fluid draining from various epithelial cells, tissues, and organs (called lymph), is collected through lymph channels and funneled to lymph nodes through their afferent lymphatic vessels. The assorted antigen-presenting cells (APC) residing in the nodes can then sample the various antigens, particles, and microbes present in the lymph fluid. In addition, dendritic cells can directly transport digested antigenic particles through lymph fluid to the nodes. Naive T and B lymphocytes enter the nodes through the arterial circulation and migrate through specialized high endothelial venules (HEVs) to reside in the node where they encounter antigens. The lymph nodes are organized into various compartments that concentrate the different lymphocyte subsets. T cells migrate to the paracortical areas of the nodes where they encounter antigens that are displayed by dendritic cells. The B cells form follicles at the periphery of the nodes that often contain germinal centers in which rapid B-cell maturation and proliferation occurs. Once activated, lymphocytes exit the lymph nodes through efferent lymph channels to reach the circulation and migrate to the periphery at sites of inflammation and infection.
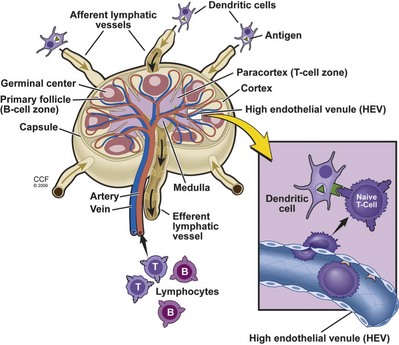
(Reprinted with permission, Cleveland Clinic Center for Medical Art & Photography © 2009-2011. All Rights Reserved.)
Cellular immune traffic is also highly organized and restricted, with certain cell populations directed to specific sites. For example, lymphoblasts and memory cells display tissue-restricted migration to extralymphoid sites such as the skin or mucosal surfaces, while lymphocytes, neutrophils, and monocytes, target and migrate to sites of inflammation in response to local mediators. Such directed movement of immune cells is termed homing, and results from the interaction of various homing receptors that recognize their complementary ligands on the surface of specialized vascular endothelium in selected tissues (Salmi and Jalkanen, 1997). Much of this directional trafficking is orchestrated by two families of G protein–coupled receptors: chemokine receptors and sphingosine-1-phosphate (S1P) receptors (Cyster, 2005). For example, in the spleen, lymphocytes enter the lymphoid area (white pulp) from the arterioles, pass to the sinusoids of the erythroid area (red pulp), and leave by the splenic vein.
Dendritic Cells
Dendritic cells originate in the bone marrow but are dispersed and populate the epithelium of common sites of entry for microbes, such as the respiratory and gastrointestinal tracts and the skin. In the skin, epidermal dendritic cells are called Langerhans cells. They are also dispersed throughout most solid organs. They are characterized by thin membranous projections, which are helpful for capture of large protein antigens. Dendritic cells are commonly referred to as “professional antigen-presenting cells (APCs),” because they display antigens and provide activating signals to T and B lymphocytes. Dendritic cells not only capture antigens, but also transport them to peripheral lymphoid tissues where they encounter lymphocytes. During their migration to lymph nodes, dendritic cells mature by increased synthesis and expression of MHC and surface costimulatory molecules needed for T-cell activation (Villadangos and Schnorrer, 2007). In addition, dendritic cells play a central role in the differentiation of naive T cells into their various subsets.
Lymphocytes: T and B Cells
T lymphocytes originate in the bone marrow and migrate to the thymus where they undergo a complex process of maturation. Mature T cells are responsible for cell-mediated cytotoxicity, providing help to antibody-producing B cells, delayed-type hypersensitivity, and play specific roles in immune regulation. Stem cells first enter the outer cortical thymus and mature as they pass to the thymic medulla. During this migration, the critically important surface T-cell receptor (TCR) genes are rearranged, and the CD3, CD4, and CD8 surface molecules appear (Starr et al, 2003). During maturation CD4−CD8− (double-negative) T cells develop into CD4+CD8+ (double-positive) T cells, and then become single-positive CD4−CD8+ (cytotoxic/suppressor) or CD4+CD8− (helper/inducer) T-cell precursors. Each retains the pan–T-cell CD3 marker, which is a signal-transducing component of the TCR. Thymic T-cell maturation also involves the critical initial process of self-recognition, which is a consequence of the interaction between maturing T cells and the surrounding thymic cortical epithelium. This leads to the selection of those T cells that are to be saved. Negative selection occurs when CD4+CD8+ TCR+ thymocytes recognize self-peptide–MHC complexes on thymic epithelium with high avidity, which leads to apoptosis or deletion of these clones. The majority of cortical thymocytes are killed during the selection process, thus preventing the development of autoimmune T cells. During positive selection CD4+CD8+ TCR+ thymocytes recognize self-peptide–MHC complexes on thymic epithelial cells with low avidity. This saves them from programmed cell death. The resulting T-cell populations are self-MHC restricted and tolerant to self-antigens, but are capable of reacting to foreign antigens. Cells leaving the thymus migrate to the peripheral lymphoid organs and seed the T-dependent regions of lymph nodes, spleen, and lymphoid follicles (Przylepa et al, 1998).
Trafficking of Naive T Cells
Rather than remain passively in the lymphoid tissues, naive or antigen inexperienced T cells circulate through the secondary lymphoid tissues using the lymphatic and blood vessels (Picker and Butcher, 1992). From the blood vessels, T cells enter secondary lymphoid tissues using specialized carbohydrate binding proteins termed selectins. Naive T cells express high levels of L-selectin (CD62L) that mediates binding to its ligand, peripheral lymph node vascular addressin (PNAd). PNAd is expressed by specialized postcapillary venules, called high endothelial venules (HEV), draining the lymphoid node. CD62L-PNAd engagement ceases the circulation of the T cells, allowing the T cells to traverse the HEV barrier and enter the cortical areas of the lymph node. The cortical areas of lymphoid tissues are rich in professional antigen-presenting cells such as macrophages and dendritic cells (see Fig. 17–3). As the naive T cells percolate through this area the T-cell receptors on each cell can survey the surface of these APCs for recognition of the specific peptide-MHC ligand, leading to cellular activation. This restricted circulation of naive T cells optimizes the chance of T-cell encounters with dendritic cells presenting the specific ligand, and is critical for efficient immune surveillance in light of the low number of T cells specific for any particular antigen-MHC complex (approximately 1 in every 105 to 106 cells). Unless the T cell encounters the appropriate activating complex presented by an antigen-presenting cell, the naive T cells exit the lymph node through the efferent lymphatics and are eventually carried into the thoracic duct. The thoracic duct drains into the blood vessels, and the pattern of naive T-cell circulation through the secondary lymphoid tissues continues.
Molecules of the Adaptive Immune System
Major Histocompatibility Complex
Both class I and II molecules are dimers with an α and a β chain (Fig. 17–4). The class I alpha chain has three domains of amino acids and a constant β chain called β-2 microglobulin, which is the same in all class I molecules and does not insert into the cell membrane (Bjorkman, 1987). The class II molecules have two domains for each chain, and both chains insert into the cell membrane. The alpha-1 and -2 domains of class I molecules and the alpha-1 and beta-1 domains of the class II molecules form a unique antigen peptide–binding cleft that cradles the antigen and is essential for antigen presentation. Analysis of peptides eluted from the binding groove of MHC class I molecules revealed that a maximum of 8 to 10 amino acids will fit, while the groove for the MHC class II molecule is larger and allows a peptide of 12 to 28 amino acids in length. The β-2 microglobulin domain of the class I and the alpha-2 and beta-2 domains of the class II molecules are fairly invariant, so the diversity of the different HLA antigen molecules resides in the domains that define the peptide-binding groove.
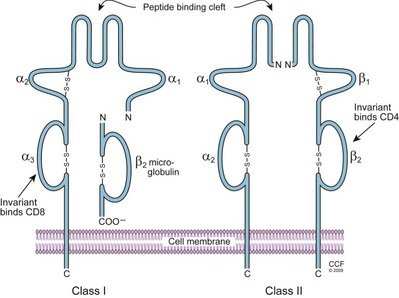
(Reprinted with permission, Cleveland Clinic Center for Medical Art & Photography © 2009-2011. All Rights Reserved.)
T-Cell Receptor
The TCR is that cell surface structure responsible for the initial steps in T-cell activation upon encounter with antigen. The TCR comprises at least seven receptor subunits whose production is encoded by six separate genes that are precisely assembled (Davis and Bjorkman, 1988). The TCR is composed of the clonotypic α and β chains that are responsible for antigen binding and are noncovalently associated with the invariant CD3-γ, -δ, -ε, and TCR ζ chains. The cytoplasmic tails of the CD3 and TCR ζ chains contain a critical amino acid sequence known as the immunoreceptor tyrosine–based activation motif (ITAM) that includes two tyrosine residues. The CD3 chains contain one ITAM, while the TCR ζ chain contains three ITAM sequences (Fig. 17–5). CD3 is commonly called the pan–T-cell marker, present on all T cells. There are generally two types of TCRs named TCR1 and TCR2. The TCR1 consists of a γδ heterodimer, appears first in ontogeny, and represents about 5% of circulating T cells in an adult. The TCR2, representing 95% of peripheral blood T cells, has an αβ heterodimer. Each of the two polypeptides comprising each receptor has a constant and a variable region (similar to immunoglobulin). Antigen recognition takes place at the variable αβ heterodimer, resembling an antibody Fab antigen–binding fragment. The relatively invariant transmembrane CD3 complex transmits the signal to the cell interior. Reminiscent of the diversity of antibody molecules produced by rearrangements of variability, diversity, and joining genes, the TCRs can likewise identify a large number of antigenic specificities, estimated to be 1010 to 1012 epitopes (Davis et al, 1998). The affinity of TCR binding to APCs is rather weak, which is why additional adhesion molecules are required to strengthen the interaction.
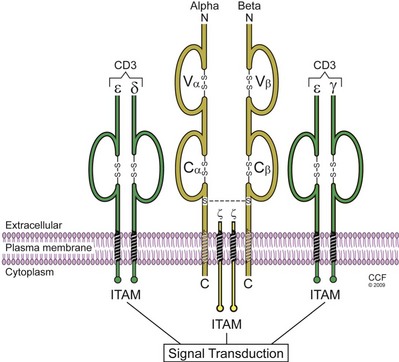
 ) with signaling functions termed immunoreceptor tyrosine based activation motifs (ITAMs). The phosphorylation of the tyrosines in ITAMs represent the initial intracellular signaling events induced by TCR interaction with antigen.
) with signaling functions termed immunoreceptor tyrosine based activation motifs (ITAMs). The phosphorylation of the tyrosines in ITAMs represent the initial intracellular signaling events induced by TCR interaction with antigen.(Reprinted with permission, Cleveland Clinic Center for Medical Art & Photography © 2009-2011. All Rights Reserved.)
Lymphocyte Activation
Presentation of Antigen
An essential requirement of immunity is that the system cannot be continuously triggered by self-antigens. As described previously, an early process of selection of T cells during maturation in the thymus deletes many offending clones, and selects those for future antigen response by utilizing MHC restriction. The TCR2 T cells bearing αβ receptors only respond when the antigen-presenting cells express the same MHC molecules as the host from which the T cells were derived (the MHC molecules that first primed the T cell). The types of responses are also controlled by the specificity of the MHC antigens on the APCs themselves. Those T cells bearing CD8 that are programmed to be cytotoxic require APCs to present antigen in the context of MHC class I molecules, while those helper T cells expressing CD4 require antigen to be associated with MHC class II molecules on the APC for activation (Micelli and Parnes, 1993).
An additional layer of control is also imbedded in this system. Antigen must be processed into smaller pieces, linear 8 to 20 amino acid sequences, so it can fit precisely into a groove created by the outermost domains of the class I and class II MHC molecules (Germain, 1994). The result is that the T cell must recognize both MHC and peptide in a particular arrangement for antigen-specific activation to take place (Madden, 1995). Antigen processing necessitates the conversion of native proteins to peptide-MHC complexes. This involves uptake by the cell, proteolysis, and additional mechanisms for the repackaging and transport of the MHC-peptide complex to the cell surface. In general, peptides associated with MHC class I are derived from proteins synthesized within the cell on cytosolic ribosomes, such as viral and oncogenic products, whereas those peptides associated with MHC class II molecules often originate from extracellular fluids, such as bacterial products and toxins, and are taken up by receptor-mediated endocytosis.
Antigen-Presenting Cells
An antigen-presenting cell (APC) is a cell that can process a protein antigen, breaking it into peptides and then presenting it in conjunction with MHC molecules on the cell surface, where it may interact with appropriate T-cell receptors. Macrophages, monocytes, some B cells, Langerhans cells of the skin, dendritic reticulum cells, and vascular endothelium can process and present antigen. The most efficient are the dendritic cells, which have a characteristic starfish shape and line the epithelial layer of exposed surfaces. Once mature, they travel to the periphery to pick up antigen, then process and present it to naive T cells in the lymphoid tissues. These cells ingest proteins and split them into small peptides in endosomes. The peptides are then transported to the cell surface and assembled in the peptide-binding groove on the MHC molecules for presentation (Trombetta and Melman, 2005). The peptides presented by APCs must be assembled with self-MHC molecules, forming the basis of MHC restriction in the immune response. CD4+ T cells will recognize antigens only in the context of self-MHC class II molecules on the surface of the APCs. Again, cells that can initiate an immune response, such as dendritic cells, macrophages and some B cells, are often referred to as “professional APCs.”
Three Signals
T cells become activated when the TCR engages an antigen for which it is specific; however, the TCR must “see” the antigen peptide presented in the groove of an MHC molecule on an APC. The interaction of one TCR and a peptide-MHC is of surprisingly low affinity and must be stabilized by an appropriate coreceptor–CD4 for MHC class II and CD8 for MHC class I molecules (Fig. 17–6). This initial step is called signal 1. Although triggering by signal 1 is necessary, it is not sufficient for T-cell activation alone. In fact, stimulation by signal 1 alone leads to a state of anergy, whereby the T cell becomes unresponsive to antigen. Activation requires costimulation by proteins on the APC that engage specific receptors on the T-cell surface, referred to as signal 2. The major APC molecules providing signal 2 are B7.1 (CD80) and B7.2 (CD86), but a number of other accessory molecules may support or enhance costimulation, such as CD2 engaging LFA-3 (CD58) and LFA-1 engaging ICAM. These molecules engage the T-cell surface glycoprotein CD28, and when combined with signal 1, leads to activation of calcium-calcineurin phosphatase, mitogen-activated protein (MAP) kinase, and protein kinase C pathways, which in turn initiate transcription (Dustin, 2005) (see Signal Transduction). Completion of activation requires the delivery of growth factors (interleukin [IL]-2 and -15), using phosphoinositide-3-kinase (PI-3K) and the target of rapamycin (mTOR) pathways, referred to as signal 3, thus initiating the cell cycle.
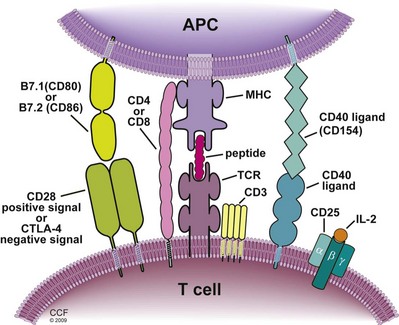
Recognition of Alloantigen
The immune response is provided by two distinct pathways for allograft recognition (Gould and Auchincloss, 1999). In the direct recognition pathway, recipient T cells engage peptide-MHC molecules expressed on allogeneic cells. The direct pathway is mainly responsible for the initiation of acute cellular rejection of allografts, and evidence suggests that passenger leukocytes (dendritic cells) play a key role. These donor APCs must express both MHC class II and the appropriate costimulatory molecules, because the depletion of either will prevent an immune response from taking place. The peptides sitting in the MHC groove may be derived from endogenous proteins of either the donor or the recipient. Thus the foreign MHC molecules presented to the recipient can be a myriad of alloantigens, including the allogeneic MHC molecule itself plus the many different peptides derived from endogenous proteins. There is also evidence that recipient T cells can recognize allo-MHC molecules that are empty, with no peptide in the binding groove. These observations help explain why so many recipient T cells can become alloreactive and rapidly destroy a graft.
Lymphocyte Precursor Frequency
The magnitude of the responses produced by various subsets of lymphocytes is closely related to the absolute number of precursor cells capable of responding. Thus the frequency of precursor cells can determine the robustness of an antigen-specific response in culture. Simply counting cells with a particular marker (i.e., CD4) may over- or underestimate the number of cells able to perform the function associated with cells bearing that marker. So, a technique called limiting dilution analysis (LDA) is used to measure the abundance of cells able to perform a particular function. The particular function can be CD4+ helper-cell proliferation measured by 3H-thymidine incorporation, or CD8+ CTL lytic activity of chromium-labeled targets. The LDA procedure involves setting up cell cultures with graded dilutions of cell suspensions (peripheral blood lymphocytes) to be tested. If the quantity of active cells in the suspension is so high that each culture is positive for the specificity being tested, then all the cultures will be positive. If the concentration of cells is very low, then only rarely will a positive culture be found. Between these extremes, the response can be quantified as a function of cell dose. The analysis follows a Poisson distribution in that the dilution at which 37% of the aliquots give negative responses will contain one precursor cell per aliquot. From this concentration one can calculate the precursor frequency for the original cell suspension (Lefkovits and Waldmann, 1979).
Regulation of T-Cell Activation
Until they are activated through TCR-mediated engagement of a specific antigen-MHC complex on professional APCs, T cells are immunologically quiescent and do not express immune functions. Activation of specific T cells and the generation of an immune response require transport of antigenic components to the lymphoid tissue. For the induction of most T-cell–mediated immune responses, the critical antigen-presenting cell is the dendritic cell, which is interspersed throughout peripheral tissues (Shortman and Naik, 2007). As situated in the peripheral tissues, dendritic cells exist in an immature state and must be activated to perform the immune functions of antigen processing, presentation, and trafficking to the lymphoid tissue.
The immune system has evolved with highly regulated components to control the activation of dendritic cells and the transport of antigenic material to the lymphoid tissue. Tissue trauma and/or the deposition of antigenic material in peripheral tissues, such as from a microbial infection, vaccination, or an organ allograft, induces a local inflammatory response. This inflammation is initiated by the production of TNF-α and IL-1 in response to tissue trauma and/or recognition of microbial products (Tracey and Cerami, 1994). Microbial products such as flagellin, unmethylated CpG of DNA, lipopolysaccharide, or single- or double-stranded RNA are potent activators of cells through innate pattern recognition and Toll-like receptors. In addition, endogenous stress molecules released by a variety of cells during tissue injury include HMGB1, heparin sulfate, and hyaluronan fragments bind to TLR2 and TLR4 to activate cells. An important consequence of this response is that inflammation is stimulatory for antigen capture, processing, and transport to the lymphoid tissues. Interstitial dendritic cells are activated by TNF-α to become phagocytic cells and ingest pieces of foreign material and tissue debris. The ingested material is processed or degraded within specialized intracellular compartments, and peptides and glycolipids are presented as complexes in association with class I or class II MHC–encoded proteins. TNF-α produced during tissue inflammation also stimulates the dendritic cells to downregulate the expression of molecules retaining or tethering dendritic cells in parenchymal tissues, and to upregulate expression of receptors for chemoattractant cytokines, or chemokines, that direct trafficking of the dendritic cells through the afferent lymphatic vessels and into the T-cell rich areas of lymphoid tissues draining the tissue site (Saeki et al, 1999).
Following trafficking to the lymphoid tissue, dendritic cells presenting peptide-MHC complexes secrete proteins that attract naive T cells to survey or sample the dendritic cell surface. During antigen presentation in the lymphoid tissues, T cells expressing receptors with specificity for peptides presented in association with class II MHC molecules (i.e., CD4+ T cells) and/or T cells with specificity for peptides presented in association with class I MHC molecules (i.e., CD8+ T cells) are stimulated to develop into effector cells (Germain and Margulies, 1993). In addition to TCR engagement of peptide-MHC complexes, activation of naive T cells to functional activity requires other receptor–ligand interactions (Schwartz, 1992). This costimulation is provided by several complementary sets of molecules. During the initial period of T-cell priming, TCR engagement of peptide–class II MHC complexes on the dendritic cell stimulates the CD4+ T cell to express the ligand CD154, which binds to a costimulatory molecule, CD40, constitutively expressed by dendritic cells. Engagement of this ligand–receptor pair stimulates the dendritic cell to upregulate expression of the CD80/B7-1 and/or CD86/B7-2 costimulatory molecules that bind to a constitutively expressed molecule, CD28, on the T-cell surface.
Activation of B Cells
Antibody Production
Although the thrust of this chapter focuses on cell-mediated immunity, a brief description of antibody structure follows. Antibodies are glycoprotein molecules of the globulin fraction, produced by plasma cells in response to stimulation with an immunogen. They are also called immunoglobulins and constitute 1% to 2% of the total serum proteins. The term antigen is used to describe a molecule capable of being recognized by an antibody or a T-cell receptor. The immunoglobulin molecule consists of two identical heavy (H) and two light (L) chains held together by disulfide bonds (Fig. 17–7). There are five antibody classes termed isotypes, so named by their different heavy chains and designated IgM, IgG, IgA, IgD, and IgE. Two types of light chains, kappa and lambda, are present in all five antibody classes, although only one is present in a specific molecule. The basic structure is Y shaped with a hinge region rich in proline, stabilized by the disulfide bridges and noncovalent forces. Both H and L chains have constant amino acid sequences at their carboxy terminus and variable sequences at their amino terminus (Alzari et al, 1988).
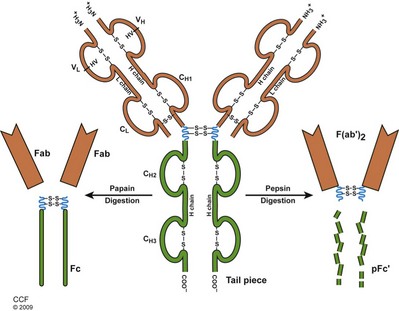
(Reprinted with permission, Cleveland Clinic Center for Medical Art & Photography © 2009-2011. All Rights Reserved.)
Much has been learned about antibody structure and binding after digestion experiments using the proteolytic enzymes papain and pepsin (see Fig. 17–7). Digestion with papain yields two Fab (antibody binding) and one Fc (crystallizable) fragment(s). Each Fab fragment has one antibody-binding site. The Fc fragment does not bind antibody, but is responsible for fixation to complement and attachment of the molecule to the cell surface. Digestion with pepsin cleaves the molecule toward the carboxyl-terminal side of the disulfide bonds, resulting in a single F(ab′)2 fragment with two antigen-binding sites and a functionless heavy chain segment. These experiments confirmed that specific antigen binding occurs at the Y limb of the molecule, and that the nonspecific functions of each antibody isotype reside in the Fc end (Harris et al, 1999).
Monoclonal Antibodies
The in vivo antibody response to an antigen is polyclonal in that many different antibody molecules are generated that will bind to the antigen. Each antibody originates from a specific clone of plasma cells, and binds to a different antigenic determinant or epitope. Monoclonal antibodies are synthesized from a single clone of cells. The identical copies produced contain only one class of H chain and one type of L chain. They are produced by fusing antigen-primed splenic B cells with an immortal myeloma cell line to create a hybridoma (Kohler and Milstein, 1975). Polyethylene glycol is used to effect the cell fusion. The antibody-producing B cell provides the specificity, and the myeloma cell provides the immortality to the hybridoma, which can generate large quantities of the monoclonal antibody. For clinical practice, murine-derived monoclonal antibodies can be engineered by “humanizing” the sequences of the constant regions of the light and heavy chains or with recombinant technology actually fusing human constant regions to murine variable regions to create a “chimeric” antibody. This is done to minimize undesired reactions of the human host to the murine sequences, which may trigger inflammatory responses such as fever, chills, nausea, vomiting, joint pains, hypotension, tremors, seizures, or visual disturbances.
Allogeneic B-Cell Activity
B cells often require the help of CD4+ T cells to proliferate and differentiate into plasma cells that synthesize and secrete specific antibody (Parker, 1993). This is especially true when B cells encounter alloantigens from an organ transplant. After B-cell immunoglobulin receptors react with donor protein antigens, the antigens are endocytosed, processed, and transported to the cell surface for presentation to resting T cells, only in the context of MHC class II molecules on the B-cell surface. The helper CD4+ T-cell TCR is engaged and stabilized by its CD4 molecule, which leads to T-cell activation and secretion of lymphokines including IL-2 (see Fig. 17–6). These lymphokines promote B-cell growth and differentiation into plasma cells that secrete specific antibodies. B-cell activation also requires the presence of a second costimulatory signal to complete the process. In this case, a marker on the B-cell surface, CD40, must be engaged by its ligand CD40L on the surface of the helper T cell (Foy et al, 1996). T cells are also required for isotype switching that occurs during some immune responses (Stavnezer, 1996). Plasma cells often secrete IgM antibodies after primary antigen exposure. Secondary responses (after reexposure to the same antigen) usually results in the production of IgG or IgA antibodies by B cells with the same antibody-binding variable regions.
Cell Signal Transduction
TCR-Signaling Proximal Events
Activation of T cells is dependent on the signaling events transmitted by the TCR to the cytoplasm and the nucleus (Fig. 17–8). The initial intracellular signaling events, after lymphocyte detection of specific antigen, begin with the CD3 molecules (CD3ε, γ, and δ) and their corresponding motifs termed ITAMs (immunoreceptor tyrosine-based activation motifs), which are phosphorylated following TCR ligation (van Leeuwen and Samelson, 1999; Kane et al, 2000; Myung et al, 2000; Smith-Garvin et al, 2009). Phosphorylation of ITAMs is mediated by two Src family kinases: Lck, which constitutively associates with CD8 and CD4 molecules, and Fyn, which interacts with the cytoplasmic domains of the CD3ε and ζ chain after receptor engagement. The phosphotyrosines produced by tyrosine kinases form binding sites for protein domains referred to as SH2 domains (Src homology 2 domains) that are present in a number of signaling molecules. The interaction of SH2 domains with the phosphotyrosines provides an important way to recruit signaling proteins to the activated receptors. TCR signaling can also be negatively regulated by the protein tyrosine kinase Csk, which alters the phosphorylation status of the Src kinases. The kinase domain of the Src kinase contains two tyrosine residues, where one provides an activation signal while the other inactivates the enzyme. In resting cells, the Csk kinase phosphorylates the inhibitory tyrosine that maintains the Src enzymes in an inactive state. The negative effect of Csk on Src kinase function can be reversed by the protein tyrosine phosphatase CD45, which is involved in receptor signaling in T cells. This involves the removal of the phosphate group from the inhibitory tyrosine residue of the Src kinase.
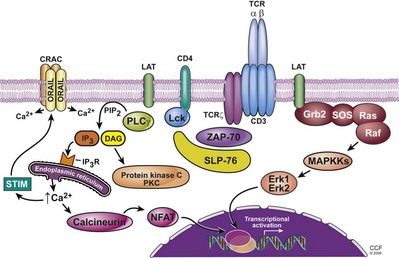
The most effective activation of the TCR requires interaction with the coreceptor molecules CD4 and CD8, which serves to bring Lck in close proximity to the cytoplasmic domains of the TCR complex (van Leeuwen and Samelson, 1999; Kane et al, 2000; Myung et al, 2000). Once activated, Lck phosphorylates the tyrosine residues in the ITAMS of the CD3 and TCR ζ chains, which then provide the docking site for the tyrosine kinase ZAP-70. After recruitment to the TCR, ZAP-70 becomes activated as a result of its phosphorylation on multiple tyrosine residues by either Lck or Fyn kinases. Activated ZAP-70 then leads to the activation of the adaptor molecule LAT (linker of activation in T cells) and SLP-76. LAT is a transmembrane protein expressed in T cells and NK cells. It is a critical SH2 domain–binding protein involved in transmitting the signal from the membrane to downstream targets. LAT is essential for coupling the TCR to the PLC-γ1–Ca2+ and the Ras signaling pathways. SLP-76 is another adaptor protein that is also important for efficient TCR-induced tyrosine phosphorylation of PLC-γ1, inositol phosphate accumulation, Ca2+ mobilization, and Ras activation. Thus the proximal signaling complex composed of the adaptor molecules along with a growing number of effector molecules is responsible for promoting the TCR–phosphotyrosine kinase (PTK) signal into multiple distal signaling pathways (Smith-Garvin et al, 2009).
Activation of Intracellular Calcium and Protein Kinase C (PKC)
Phosphorylation of the enzyme phospholipase C–γ (PLC-γ) is a critical event in transmitting the activation signal from the membrane downstream (Myung et al, 2000). After TCR ligation, PLC-γ is present in the proximal signaling complex bound to SLP-76 and LAT where it is phosphorylated, an event that is dependent on several kinases (Lck, ZAP-70, Itk, and a Tec kinase family member, Rlk). The activated PLC-γ then hydrolyzes membrane-bound phospholipid phosphatidylinositol bisphosphate (PIP2) into two active components, inositol triphosphate (IP3) and diacylglycerol (DAG).
IP3 stimulates Ca2+-permeable ion channel receptors (IP3R), resulting in the release of Ca2+ into the cytosol from storage sites within the endoplasmic reticulum (Oh-hora and Rao, 2008) which raises the intracellular levels of Ca2+. The reduction in endoplasmic reticulum (ER) Ca2+ stimulates a sustained influx of extracellular Ca2+ via the activation of plasma membrane Ca2+ release–activated Ca2+ (CRAC) channels. Recent studies have identified the sensor for the depletion of ER Ca2+ stores and the activator of CRAC as an ER transmembrane protein referred to as stromal interaction molecule (STIM) (Roos et al, 2005). Elevated µCa2+ results in the activation of multiple transcription factors along with various signaling molecules. This includes the Ca2+ enzyme calcineurin, a cytosolic serine/threonine protein phosphatase that regulates the activation of an important family of transcription factors, termed NFAT (nuclear factor of activated T cells).
NFAT transduces Ca2+ signals not only in T lymphocytes but also in B cells, NK cells, monocytes, and nonhematopoietic cells, including those of the cardiac and nervous system (Zhu and McKeon, 2000). Four separate genes are present that encode different NFATs: NFAT1/p, NFAT2.c, NFAT3, and NFAT4. NFAT, along with other transcription factors, play a critical role in T-cell activation and clonal expansion through their activation of the interleukin-2 gene. The inactive form of NFAT is retained in the cytoplasm, unable to translocate to the nucleus because its nuclear localization sequence (NLS) is phosphorylated on serine/threonine. Calcineurin is composed of a catalytic subunit and a regulator subunit and is activated by the recruitment of Ca2+-bound calmodulin. The association of calmodulin with calcineurin alters the structure of catalytic subunit, leading to activation of its phosphatase activity. Calcineurin then dephosphorylates the NLS of NFAT, allowing the transcription factor to translocate into the nucleus.
NFAT is important for T-cell activation; and because of its role in the regulation of critical genes in this process (e.g., IL-2, proto-oncogenes c-myc and cytokine receptors), it is a target for immune suppression in the allotransplant setting (Matsuda and Koyasu, 2000). The immunosuppressive drugs cyclosporine and tacrolimus function by blocking the NFAT pathway. Cyclosporine is a small, cyclic, fungus-derived peptide that is immunosuppressive once it has formed a complex with its cytoplasmic receptor, cyclophilin, a ubiquitous cytosolic protein. The activity of tacrolimus also is dependent on binding to its receptor, FK-binding protein 12 (FKBP12). The active complexes, cyclosporine/cyclophilin and tacrolimus/FKBP12, are known to block NFAT nuclear translocation by binding to the catalytic subunit of calcineurin and inhibiting its phosphatase activity. The major inhibitory activity of cyclosporine and tacrolimus is thought to be their suppression of the growth factor IL-2. Actually, the induction of IL-2 gene expression along with other T-cell activation genes involves stimulation of NFAT/AP-1 transcription factors and Ras signaling while NFAT activity in the absence of AP-1 stimulation induces a gene expression pattern that promotes T-cell anergy (Macian et al, 2002).
The cleavage of the PIP2 by the PLC-γ enzyme also results in the formation of diacylglyerol (DAG), which remains attached to the inner surface of the plasma membrane (Myung et al, 2000). DAG is involved in the activation of protein kinase C (PKC), a serine/threonine protein kinase that regulates the nuclear translocation of various transcription factors, including NFκB. Recent studies have detailed how PKC activation leads to nuclear import of NFκB via the phosphorylation of a multiple protein complex (Matsumoto et al, 2005). PKC can be further activated by the increase in Ca2+ resulting from the release of IP3. Beside activation of the PKC pathway, TCR-induction of DAG leads to activation of the Ras pathway.
Ras Pathway
Activation of the TCR-associated tyrosine kinases involves the small guanosine triphosphate (GTP)-binding protein Ras (Ebinu et al, 2000; Genot and Cantrell, 2000). Ras proteins are proto-oncogenes that were initially discovered because of their effect on tumor cell growth, but it is now known that Ras plays an important role in lymphocyte activation. Ras is involved in the activation the AP-1 family of transcription factors, which are composed of heterodimers of the oncogenes fos and jun. It also regulates other transcription factors such as NFAT, Elk-1, and the serum response factor (SRF). Ras also mediates TCR signals important to thymocyte and B-cell development. Defects in the activation of Ras have been linked to the development of T-cell anergy. Ras is a guanine-nucleotide binding protein that rapidly cycles from an inactive guanosine diphosphate (GDP) form to an active GTP-bound state. Ras activation is enhanced by two different proteins (SOS and RasGRP) and is suppressed by GTPase-activating proteins (Smith-Garvin et al, 2009). The activation of Ras induces a cascade of protein kinases referred to as the mitogen-activated protein kinases (MAP kinase). One of the best characterized Ras effector pathways is mediated by the MAP kinase Raf-1. Activation through the TCR results in the tyrosine phosphorylation of LAT that binds to Grb2 and recruits the guanine-nucleotide exchange factor SOS that activates the G protein, Ras. Activated Ras recruits Raf-1 to the plasma membrane, where Raf-1 is activated. Raf-1 then phosphorylates and activates the kinase MAPKKS, which then activates the MAPKs Erk1 and Erk2. These events ultimately lead to the activation of a variety of transcription factors.
Actin and Cytoskeletal Changes
The interaction between TCR and APCs results in the rearrangement of the actin cytoskeletal compartment that leads to polarization and activation of T cells with a reduction in cellular motility. As part of this process, there is movement of the microtubular organizing center toward the site of contact between T-cells and APC (Jacobelli et al, 2004
Related posts:
 Definitive Therapy for Localized Prostate Cancer: An Overview
Definitive Therapy for Localized Prostate Cancer: An Overview
 Tuberculosis and Other Opportunistic Infections of the Genitourinary System
Tuberculosis and Other Opportunistic Infections of the Genitourinary System
 Prostate Cancer Tumor Markers
Prostate Cancer Tumor Markers
 Core Principles of Perioperative Management in Children
Core Principles of Perioperative Management in Children
 Neuropathic Dysfunction of the Lower Urinary Tract
Neuropathic Dysfunction of the Lower Urinary Tract
 Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Ectopic Ureter, Ureterocele, and Ureteral Anomalies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


